
Abstract
Signaling by the BCR-ABL fusion kinase drives Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL) and chronic myelogenous leukemia (CML). Despite their clinical activity in many patients with CML, the BCR-ABL kinase inhibitors (BCR-ABL-KIs) imatinib, dasatinib, and nilotinib provide only transient leukemia reduction in patients with Ph+ ALL. While host-derived growth factors in the leukemia microenvironment have been invoked to explain this drug resistance, their relative contribution remains uncertain. Using genetically defined murine Ph+ ALL cells, we identified interleukin 7 (IL-7) as the dominant host factor that attenuates response to BCR-ABL-KIs. To identify potential combination drugs that could overcome this IL-7–dependent BCR-ABL-KI–resistant phenotype, we screened a small-molecule library including Food and Drug Administration–approved drugs. Among the validated hits, the well-tolerated antimalarial drug dihydroartemisinin (DHA) displayed potent activity in vitro and modest in vivo monotherapy activity against engineered murine BCR-ABL-KI–resistant Ph+ ALL. Strikingly, cotreatment with DHA and dasatinib in vivo strongly reduced primary leukemia burden and improved long-term survival in a murine model that faithfully captures the BCR-ABL-KI–resistant phenotype of human Ph+ ALL. This cotreatment protocol durably cured 90% of treated animals, suggesting that this cell-based screening approach efficiently identified drugs that could be rapidly moved to human clinical testing.
Introduction
Targeted BCR-ABL inhibition with imatinib has improved outcome for chronic myelogenous leukemia (CML). 1 However, the situation is starkly different for pediatric and adult Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL). 2 While continuous BCR-ABL inhibition therapy—using imatinib, nilotinib, or dasatinib— potently kills Ph+ ALL cells in vitro, these drugs induce only transient remissions in patients with Ph+ ALL, ultimately allowing relapse and giving poor long-term outcomes. Although additional chemotherapy and hematopoietic stem cell transplantation (HSCT) improve the remission rate in Ph+ ALL patients, relapses remain common, 3 with an 18- to 24-month survival rate of 64% and an even worse relapse-free survival rate. 3 Differences in primary responses to BCR-ABL kinase inhibitors (BCR-ABL-KIs) in patients with Ph+ ALL and CML remain poorly understood and cannot be attributed solely to mutations in the ABL kinase domain (BCR-ABLMUTANTS). 4
We previously developed a murine leukemia model of Ph+ ALL, using primary, polyclonal Arf −/− p185BCR-ABL luciferase-expressing (Luc+) leukemia-initiating pre–B cells (LICs), which captures key genetic features of human Ph+ ALL (i.e., BCR-ABL expression and CDKN2A deletion) and enables in vivo luminescent imaging to monitor response to therapy. 5 Such BCR-ABLWT LICs retain phenotypic and signaling properties essential for suspended leukemic blasts to interact with the surrounding host environment 6 not typically present in established leukemic cell lines and remain responsive to cytokines implicated in murine pre–B-cell development both in vitro and in vivo. 6 These polyclonal BCR-ABLWT LICs can very efficiently initiate leukemias in syngeneic, immune-competent mice. 6
In “recipient” host mice engrafted with these LICs, residual BCR-ABLWT cells 5 evade treatment with BCR-ABL inhibitors, paralleling what is seen in human Ph+ ALL patients. 4 BCR-ABL kinase domain (KD) mutations, including BCR-ABLT315I (resistant to imatinib, dasatinib, and nilotinib), emerge only after long-term exposure to BCR-ABL-KIs.5,7 In contrast to the in vivo setting, BCR-ABLWT murine and human cells are quite sensitive to BCR-ABL-KIs in vitro. 8 This disparity between in vitro and in vivo sensitivity implicates the host microenvironment in attenuating the therapeutic response to BCR-ABL kinase inhibition. We previously reported that common γ chain receptor host cytokines modulate primary imatinib resistance, 6 and other host-derived growth factors have been shown to attenuate BCR-ABL kinase inhibition.9–11 Currently, no consensus exists on the relative importance of host cytokines or growth factors that attenuate the cellular responses to BCR-ABL–targeted agents.
We used a naive screening approach to identify interleukin (IL)–7 as the most potent among candidate host growth factors in inducing resistance to BCR-ABL-KIs in Ph+ ALL. Following on that study, we used a repurposing screen of existing drugs and clinical candidates to identify drugs that blocked proliferation of BCR-ABLWT LICs cultured with murine IL-7. This study identified both the majority of known antileukemic drugs and a few novel agents that could overcome cytokine-dependent cell survival signaling. Testing of combinations of these validated hits with dasatinib revealed that the antimalarial drug dihydroartemisinin (DHA) provides strong synergy with BCR-ABL-KIs against Ph+ ALL cells responding to the tumor microenvironment. When used in vivo, DHA-dasatinib combination therapy eradicates dasatinib-refractory leukemia in vivo, providing near-complete long-term survival.
Materials and Methods
Development of LIC-Based Screening Assay
Before use in any in vitro assay, LICs had to pass the following quality control parameters: test thawing (benchmark: 95% viability at 24 h), PCR genotyping to confirm Arf −/− status, mycoplasma testing (negative by Takara Bio Mycoplasma PCR assay [Clontech, Madison, WI]), sequence verification of the BCR-ABL allele, and in vitro dasatinib potency confirmation.
The maximum DMSO concentration tolerated by LICs in culture was determined to be 0.2% (by volume) and was maintained at ≤0.1% (by volume) in all subsequent work. LICs were plated in 384-well microplates at a cellular density of 5 × 104 per milliliter (1250 LICs per well in 25 µL BCM10) and confirmed to give exponential growth during the first 72 h (
Forward Cytokine Phenotypic Screen
Lyophilized murine and human recombinant cytokines (R&D Systems, Minneapolis, MN) were diluted with 1 mg/mL bovine serum albumin (BSA) in sterile phosphate-buffered saline (PBS) to a final concentration of 20 µg/mL and stored at 4 °C. Antileukemia drugs, BCR-ABL-KIs, and non–BCR-ABL–specific KIs (LC labs; Sigma, St. Louis, MO) were solubilized in DMSO to obtain 10-mM stock concentrations. Subsequent serial titrations were prepared using DMSO in 0.5-mL vials or 384-well drug master plates and stored at −20 °C.
To study the impact of murine cytokines on dasatinib action, LIC stocks containing different concentrations of candidate cytokines were prepared in multichannel sterile reservoirs and plated into 384-well plates. Drug delivery to cell suspensions was accomplished with a V & P Scientific (San Diego, CA) pin tool (
High-Throughput Phenotypic Drug Screening
The IL-7 concentration of 0.85 ng/mL was confirmed (cytokine titration assays similar to
Fig. 1a
for dasatinib) to confer near-maximal and reproducible resistance against nilotinib and imatinib in BCR-ABLWT LICs (
Fig. 1b
and
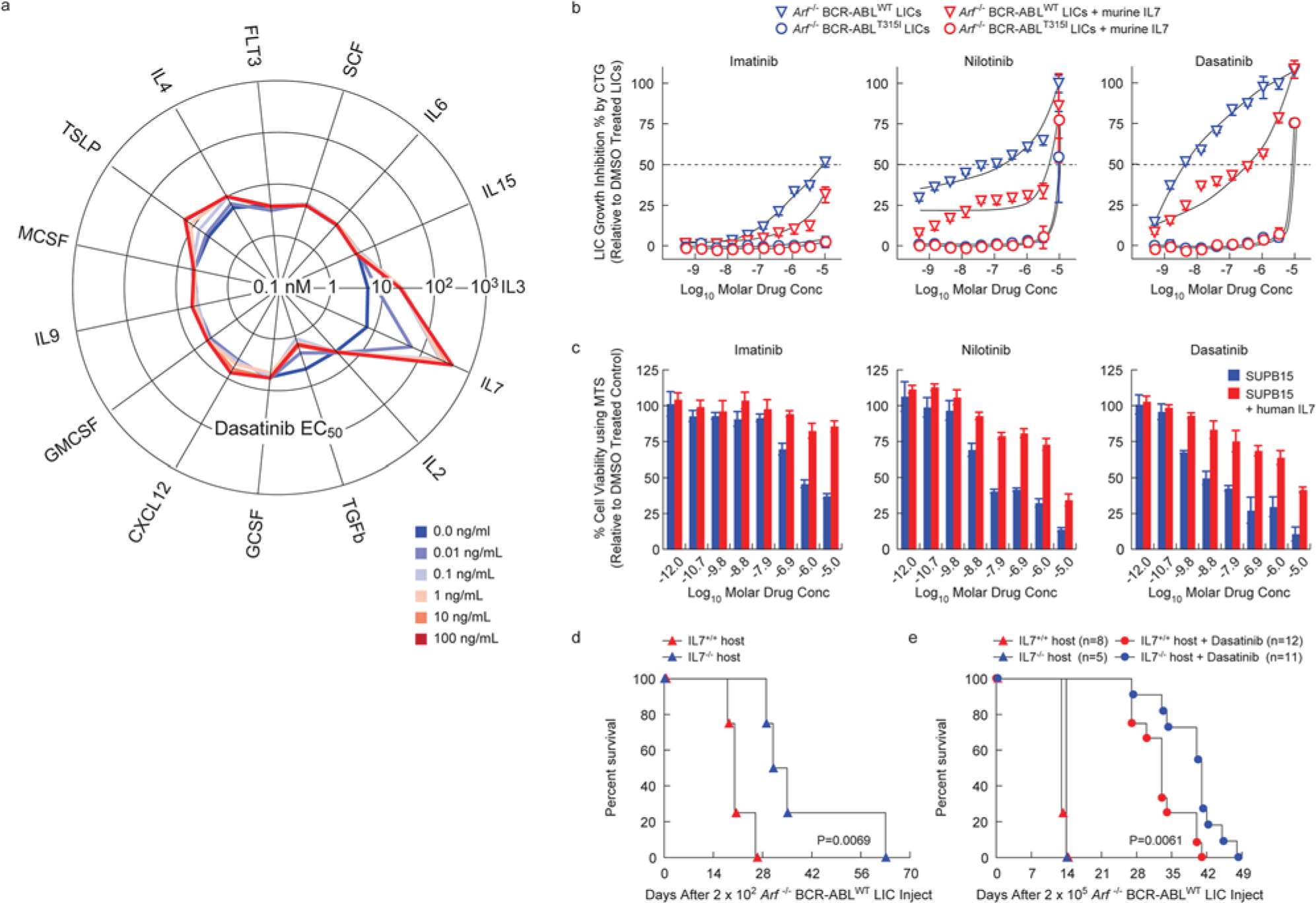
Interleukin 7 (IL-7) induces resistance to BCR-ABL kinase inhibitors (BCR-ABL-KIs) in Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL) without mutation of BCR-ABL. (
Supplementary Data
Supplementary data include (1) supplemental information (PDF), which contains materials and methods for generation of LICs, cytokine screening, fluorescence-activated cell sorting (FACS) assessment of viability and cell cycle percentages, detailed description of chemical library screened, data processing, quality control and hit scoring criteria for high-throughput (HT) drug screens, cluster analysis using therapeutic drug classes,
Results
Determining Which Leukemia Microenvironment Cytokines Induce Drug-Resistance against BCR-ABL-KIs
In the absence of drug treatment, both Arf −/− p185WT and Arf −/− p185T315I pre–B cells (referred to as BCR-ABLWT LICs
12
and BCR-ABLT315I LICs, respectively) have identical growth properties in vitro (
IL-7 provided the strongest protection against dasatinib (
Fig. 1a
). In addition, IL-3, thymic stromal lymphopoietin (TSLP, IL-7–like cytokine), and IL-4 conferred some resistance but with greatly reduced potency. In contrast, transforming growth factor–β (TGF-β) enhanced dasatinib activity. In addition, IL-7 significantly attenuated the potency of all Food and Drug Administration (FDA)–approved BCR-ABL-KIs against murine BCR-ABLWT LICs (
Fig. 1b
) and the human Ph+ ALL cell line SUP-B15 (
Fig. 1c
). However, IL-7 did not affect the potency of tested non–BCR-ABL–targeted drugs (
Physiological Levels of IL-7 Confer an Aggressive, Dasatinib-Resistant Phenotype to Ph+ ALL In Vivo
To confirm the physiological relevance of this finding, we studied the effect of IL-7 on leukemia progression and dasatinib responsiveness in vivo. Implantation of IL-7+/+, but not IL-7−/−, host mice with relatively small numbers of BCR-ABLWT LICs (200, low-burden model) led to decreased survival ( Fig. 1d ), suggesting that murine Ph+ ALL progression accelerates in response to IL-7. Injecting higher numbers of LICs (200,000) overcame this trend (200,000, high-burden model; Fig. 1e ), suggesting the phenomenon is most important during expansion of the initial seeding leukemia cell population. Treatment of animals in the high-burden model revealed a significantly shortened survival benefit from dasatinib monotherapy of BCR-ABLWT–driven leukemias in IL-7–deficient hosts ( Fig. 1e ). From a therapeutic perspective, these findings show that in vivo responses to dasatinib therapy in Ph+ ALL are modulated by IL-7, independent of BCR-ABL KD mutations. These studies also suggest that using the BCR-ABLWT LICs in the presence of IL-7 as a screening system could identify physiologically relevant pharmacological approaches to reversing this cytokine-dependent drug resistance.
High-Throughput Screens against Primary (IL-7–Induced) and Acquired (BCR-ABL Mutation) BCR-ABL-KI Drug-Resistant Phenotypes
We hypothesized that pharmacologic agents could be identified that would overcome IL-7–induced BCR-ABL-KI resistance. A collection of 5600 compounds highly enriched for approved drugs, development candidates, and compounds of known cellular activity from a variety of signaling mechanisms, including all active ingredients from approved oncology and anti-infective drugs, was screened against BCR-ABLWT and BCR-ABLT315I LICs in the presence of IL-7 (0.85 ng/mL) at a fixed drug concentration (10 µM). The quality of the screen and minimum significant activity (>10% growth inhibition) were confirmed by receiver operating characteristic analysis and other statistical metrics (
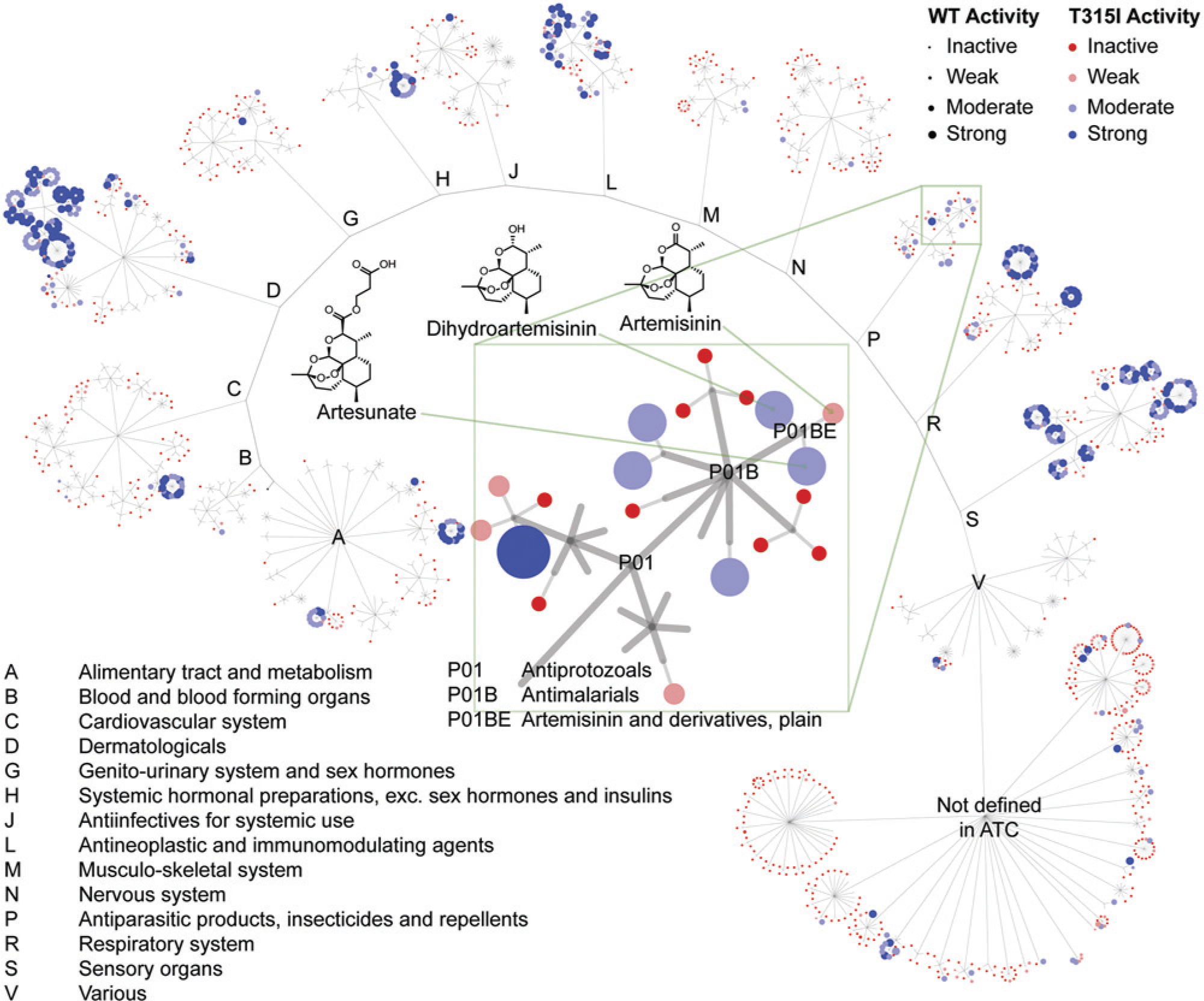
Screen to identify compounds preventing growth of wild-type and BCR-ABLMUTANT Philadelphia chromosome–positive acute lymphoblastic leukemia cells in the presence of interleukin 7. Several hundred validated hits were identified. The hits are shown as a graph with clustering hits using ATC therapeutic classification (http://www.whocc.no/atc/structure_and_principles/). A single compound can be represented more than once because of multiple therapeutic indications, as is the case for the corticosteroids (highly active clusters in A, C, D, H, R, and S). This demonstrates that the active compounds come from drugs used to treat many indications. See supplemental materials and methods and
In Vitro and In Vivo Anti–Ph+ ALL Activity of DHA
Although our validated hits included compounds with varying mechanisms of action and compounds in varying stages of development, we focused on approved drugs with favorable human pharmacokinetic and toxicity profiles that have previously been used with pediatric populations but not for leukemia, which quickly narrowed focus to the artemisinin class of antimalarials (ARTs), which includes artemisinin, artesunate, and DHA. 14 As a class, the ARTs were equipotent against BCR-ABLWT and BCR-ABLT315I LICs ( Fig. 3a ), but the anti-LIC potency of artemisinin (ca. 1 µM EC50) was weaker than that of artesunate or DHA (ca. 200 nM EC50). DHA and artesunate also killed the human Ph+ ALL cell line SUP-B15 with similar potency in the presence of IL-7 ( Fig. 3a , b ). Studies have shown that DHA kills other genotypes of leukemic cells while selectively sparing normal human lymphocytes.15,16 Therefore, DHA was chosen for further analysis because of its potency, cellular selectivity, oral formulation, favorable pharmacokinetics, known bioavailability in the hematopoietic system, and tolerability in all human age groups.
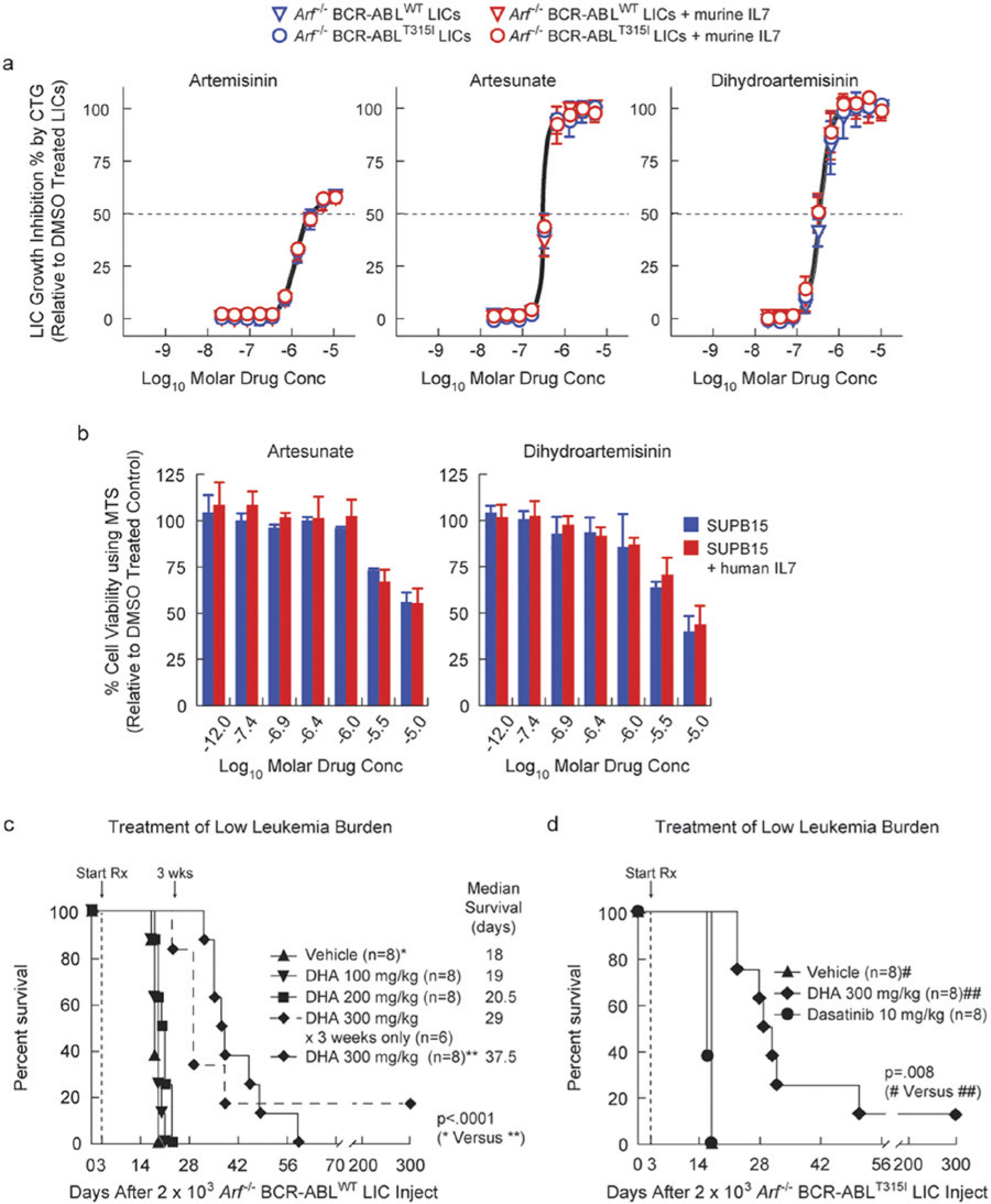
The artemisinin family of antimalarial drugs circumvents interleukin 7 (IL-7)–induced resistance to dasatinib in Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL) cells in vitro and Ph+ ALL in IL-7+/+ host mice. (
Due to our interest in modeling the approved drug DHA, which is given orally for malaria, follow-up studies in vivo used the oral route rather than the parenteral route more commonly used in oncology studies. DHA monotherapy conferred a significant dose-dependent survival benefit to IL-7+/+ host mice bearing low-burden BCR-ABLWT leukemia ( Fig. 3c ), although this required escalation to high doses (300 mg/kg) for maximal effect. During these studies, no significant changes in body weight were observed. This dose falls within the tolerated and bioavailable doses used in prior animal modeling for malaria. 17
By conventional allometry, this dose corresponds to roughly 45 mg/kg in a human, which is 4-fold higher than the highest dose used for malaria treatment in humans (up to 12 mg/kg). The limited murine pharmacokinetic data for DHA 18 indicates that it is more rapidly cleared in mice and has lower bioavailability than in humans, suggesting that lower relative doses would be efficacious in humans. In addition, existing human and murine pharmacokinetic data indicate that plasma concentrations that should be achieved in this model would be well above concentrations efficacious in vitro against LICs, even at doses more closely aligned with those used for malaria. 19 BCR-ABL mutations, which are often selected with dasatinib treatment in this model,5,7 were absent in leukemic samples collected from terminally moribund DHA-treated mice. DHA monotherapy also gave a similar response in mice with low-burden BCR-ABLT315I leukemia, which is completely refractory to dasatinib ( Fig. 3d ). Thus, unlike BCR-ABL-KIs, DHA acts against Ph+ ALL via a BCR-ABL kinase-independent mechanism.
DHA Synergistically Enhances Response to Dasatinib
Because BCR-ABLWT leukemias in IL-7–deficient mice were more sensitive to dasatinib ( Fig. 1e ), and DHA displayed anti–Ph+ ALL activity in IL-7+/+ host mice ( Fig. 3c , d ), the potential for synergy between the two drugs was evaluated. Cohorts of IL-7+/+ immune-competent mice bearing high burdens of Arf−/− BCR-ABLWT –driven leukemia were treated with dasatinib alone (10 mg/kg), DHA alone (200 or 300 mg/kg), or binary combinations ( Fig. 4a ). Despite continuous monotherapy, dasatinib induced only weak initial responses, and all animals later relapsed ( Fig. 4b ) with complete mortality within 6 weeks of initiating therapy ( Fig. 4a ). DHA alone was completely ineffective (200 mg/kg) or poorly effective (300 mg/kg) in the high-burden leukemia model ( Fig. 4a ). However, the combination of DHA and dasatinib provided a strong and rapid initial response and led to significantly increased overall survival ( Fig. 4a , b ) in comparison to the treatment with either of the two drugs alone. Of the 12 mice given 200 mg/kg DHA and 10 mg/kg dasatinib, 3 survived long term. All mice given 300 mg/kg DHA and 10 mg/kg dasatinib (n = 12) survived the treatment period. As discussed above, the plasma concentrations expected to be reached with such doses are likely providing exposure well above that efficacious against LICs and within those that could be reached in humans with good tolerability.17,19
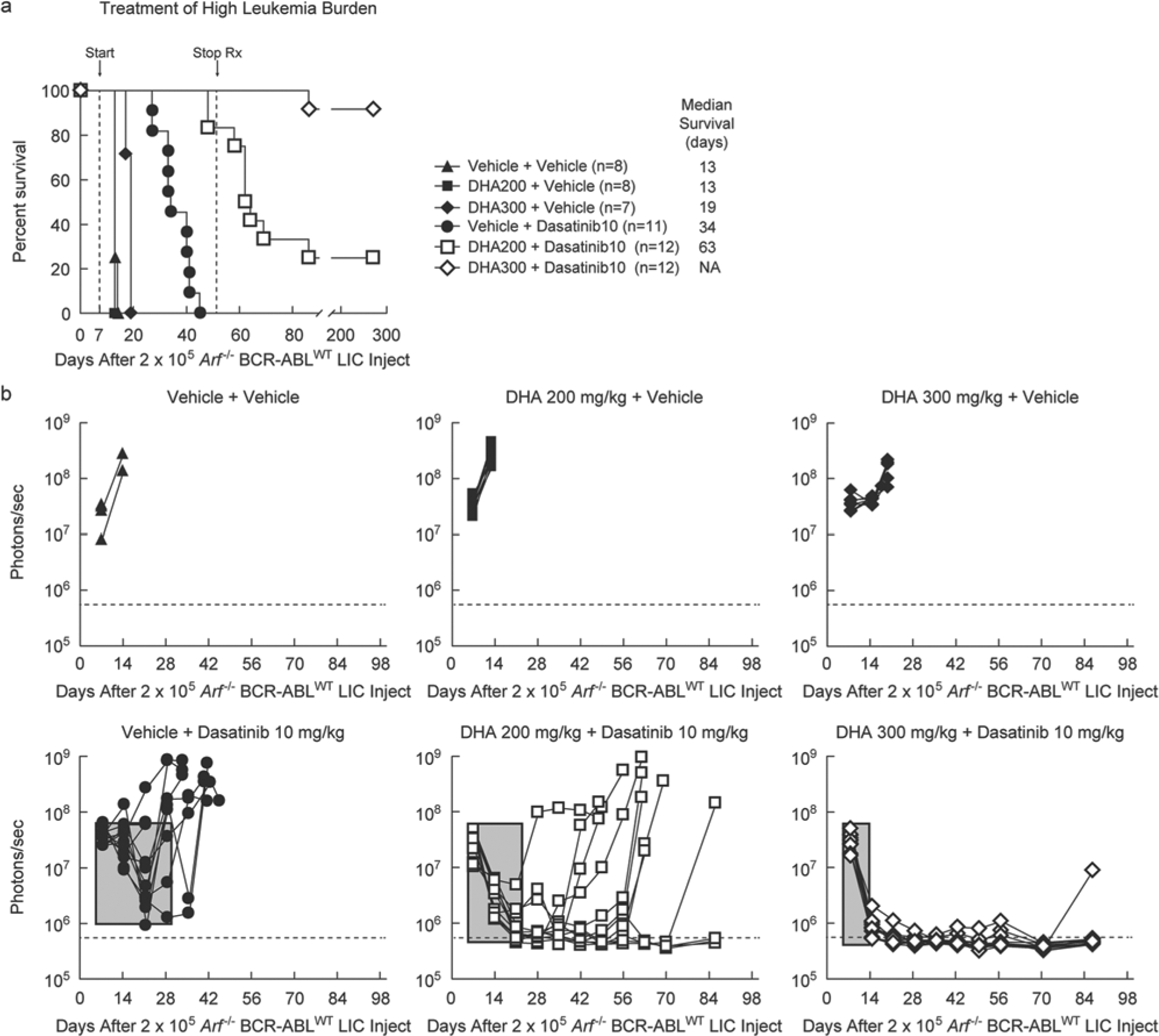
Dihydroartemisinin works synergistically with dasatinib to cure Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL). (
Four weeks after completion of combination therapy, one mouse succumbed to an isolated CNS relapse, which may be related to the poor CNS availability of dasatinib.
20
Remarkably, the 14 surviving mice taken off dasatinib-combined therapy (3/12 at 200 mg/kg and 11/12 at 300 mg/kg DHA) showed no leukemia for 1 year, the longest period monitored. In this leukemia model, all animals left untreated succumbed to full-blown leukemia within 4 weeks of injecting very low numbers of LICs (
Discussion
We identified IL-7 as the dominant (most potent) cytokine that confers resistance to all FDA-approved BCR-ABL inhibitors. IL-7 is essential for murine T-cell and B-cell development, 21 has recently been implicated in human postfetal B-cell development,21,22 and is a major contributor to the regulation and maintenance of mature T-lymphoid cells. 23 IL-7–rich niches such as bone marrow harbor early persistent drug-refractory leukemias in dasatinib-treated mice, at which point the vast majority of leukemic cells do not harbor BCR-ABL KD mutations. 5 Ultimately, BCR-ABL mutations emerge under continuous drug exposures,5,24 causing secondary drug resistance. Our findings suggest that IL-7 maintains leukemic cell viability, probably through induction of phosphorylation of STAT5, despite continuous BCR-ABL inhibition in vivo, thereby facilitating the subsequent acquisition of drug resistance–conferring kinase domain mutations. There is a well-appreciated relationship between phosphorylation of STAT5 and resistance to imatinib in cells.25–27 In addition, human Ph+ ALL patients with increased levels of pSTAT5, whose phosphorylation is triggered by IL-7, respond poorly to imatinib therapy. 27
The demonstration of IL-7–induced drug resistance provided a rationale for “complementing” pharmacological BCR-ABL inhibition with a second drug that induces IL-7–independent leukemic cell death. We assayed 5600 compounds enriched with approved drugs to identify such agents. Our studies confirmed the activities of the majority of known antileukemia drugs and also identified several new classes of the compounds with activity against both IL-7–cultured wild-type and BCR-ABL mutant LICs. We characterized the antileukemia activity of one such drug: the orally bioavailable, well-tolerated, antimalarial agent DHA, which selectively kills leukemic cells while sparing normal lymphocytes.15,16 Mechanistically, DHA acts independently of BCR-ABL-KIs ( Fig. 3 ). In combination with dasatinib, DHA enhanced the depth of remission induction ( Fig. 4b ) and achieved near-complete long-term survival in a robust murine model of human Ph+ ALL. It is noteworthy that the combination of DHA and dasatinib therapy is significantly more efficacious than the clinically used triple combination of dasatinib, dexamethasone, and asparaginase when assessed in the same model. 5
Distinct from its antimalarial mechanism of action, induction of cell death by ARTs in transformed cells is associated with accelerating the degradation of c-MYC. 28 c-MYC expression is downstream of activated STAT5, 29 which is maintained by IL-7 signaling, and STAT5 activation 30 and c-MYC expression31,32 are necessary to sustain BCR-ABL–transformed cells. This phenomenon has recently been implicated in other stroma-induced drug resistance 10 as well as used as an explanation for resistance to imatinib.10,33 However, we do not see changes in pSTAT5 levels in response to treatment with artemisinins (data not shown). Thus, DHA may synergize with BCR-ABL-KI therapy by accelerating the degradation of c-MYC, thereby countering the protective effects of IL-7, as has been reported by others. Alternatively, as has also been reported, DHA may affect BCR-ABL levels directly.34,35
We have shown that phenotypic screening using primary leukemia cells selected to maintain responsiveness to host cytokines allows identification of therapeutic agents or developmental compounds that hold promise for the treatment of Ph+ leukemias. This system provides an unbiased platform for the discovery of such new therapies and to evaluate the potential utility of such agents by demonstrating proof of concept in vivo using a murine model that closely recapitulates human disease. Finally, our results strongly support further study of BCR-ABL-KI/DHA combination therapy for the treatment of patients with high-risk Ph+ ALL.
Footnotes
Acknowledgements
We acknowledge Charles J. Sherr, Gerard Zambetti, and Joe Opfermann for critically reading the manuscript. We thank Chandra Savage and Branden Williams for assistance with mouse therapeutic studies, Christopher R. Calabrese and Monique Payton for assistance with animal imaging, Alexander J. Kovalic and Per Holmfeldt for assistance with in vitro growth factor screening experiments, Narendra P. Singh for suggestions on drug preparation for animal use, Armand Guiguemde and Jimmy Cui at the High Throughput Screening Center at St. Jude for assistance with high-throughput drug screening, Heather Mulder for performing BCR-ABL KD mutation analyses, Richard A. Ashmun and Jim Houston for performing flow cytometric cell sorting and analyses, and Owen Witte (UCLA) for the MSCV-BCR-ABL-IRES-GFP retroviral vector.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by an American Association for Cancer Research (AACR) Centennial Career Development Award for Childhood Cancer Research (R.T.W.), NIH 1R21NS066460-01 (R.T.W. and R.K.G.), Gabrielle’s Angel Foundation Medical Research Award (R.T.W.), NIH/NCI Comprehensive Cancer Center Core Grant CA-21765, and the American and Lebanese Syrian Associated Charities (ALSAC) of St Jude Children’s Research Hospital.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.