
Abstract
Huntington’s Disease is a rare neurodegenerative disease caused by an abnormal expansion of CAG repeats encoding polyglutamine in the first exon of the huntingtin gene. N-terminal fragments containing polyglutamine (polyQ) sequences aggregate and can bind to cellular proteins, resulting in several pathophysiological consequences for affected neurons such as changes in gene transcription. One transcriptional pathway that has been implicated in HD pathogenesis is the CREB binding protein (CBP)/cAMP responsive element binding (CREB) pathway. We developed a phenotypic assay to screen for compounds that can reverse the transcriptional dysregulation of the pathway caused by induced mutated huntingtin protein (µHtt). 293/T-REx cells were stably co-transfected with an inducible full-length mutated huntingtin gene containing 138 glutamine repeats and with a reporter gene under control of the cAMP responsive element (CRE). One clone, which showed reversible inhibition of µHtt-induced reporter activity upon treatment with the neuroprotective Rho kinase inhibitor Y27632, was used for the development of a high-throughput phenotypic assay suitable for a primary screening campaign, which was performed on a library of 24,000 compounds. Several hit compounds were identified and validated further in a cell viability adenosine triphosphate assay. The assay has the potential for finding new drug candidates for the treatment of HD.
Introduction
Huntington disease (HD) is a rare autosomal neurodegenerative disease for which currently only symptomatic treatments exist. 1 The disease is characterized by a CAG trinucleotide expansion encoding a polyglutamine (polyQ) tract in exon-1 of the huntingtin gene. In HD patients, a mutated huntingtin (µHtt) protein always contains more than 36 glutamine repeats, while the onset and severity of the disease are directly correlated with the polyQ length. 2 Pathogenesis arises mainly from increased gain of function of µHtt, as evidenced by the formation of soluble protein oligomers or insoluble aggregates with toxic properties, 3 while a decreased physiological function of wild-type Htt has also been proposed. 4 Both transcriptional dysregulation and mitochondrial dysfunction are believed to contribute to the disease, and there is evidence that the two processes are linked. 5
Htt was found to interact with more than 200 different cellular proteins. 6 Among these are several proteins involved in axonal trafficking and gene transcription. In the R6/2 mouse model expressing a polyQ containing an N-terminal fragment of µHtt, about 1.5% of all genes show transcriptional dysregulation and 75% of these are downregulated. 7 The interaction between Htt and several transcription factors or coactivators such as NF-Y, 8 p53, 9 Sp1 and TAF4, 10 and CREB binding protein (CBP) 11 seems to be mediated by the polyQ tract, which can sequester these proteins in the cytoplasm and deplete them from their normal nuclear localization. On the other hand, polyQ-expanded µHtt is no longer able to effectively bind to the response element-1 silencing transcription factor/neuron restrictive silencer factor (REST/NRSF) repressor, leading to increased nuclear localization of the protein and therefore suppression of brain-derived neurotrophic factor (BDNF) expression and other physiologically important neuron-restrictive silencer elements (NRSE)–regulated genes. 12 One of the transcriptional pathways that has been shown to be affected by the HD mutation in preclinical models is the cAMP pathway and associated nuclear effectors such as the leucine zipper family member CREBP and its coactivator CBP. 13 It has been shown that the inhibitory effects of µHtt on the histone acetyltransferase CBP can be counteracted by treatment with small-molecule histone deacteylase (HDAC) inhibitors in Drosophila model systems. 14 However, it was recently demonstrated that the deacetylase Sirt1 also has a protective role in several mouse models expressing µHtt. 15 An early event in HD pathogenesis is the downregulation of the cAMP responsive element (CRE) signaling pathway by µHtt,16,17 which contributes to an array of neuropathological conditions. 18 This can be mediated by direct sequestration of CBP and/or downregulation of its activity by µHtt19,20 as well as by µHtt-mediated interference with the activity of other key factors in the pathway, such as TORC1. 10 Therefore, specific aspects of the transcriptional dysregulation represent potential therapeutic targets in HD, suggesting in particular that the upregulation of CREB-mediated gene expression may be a promising approach for interfering with HD pathology.
Several primary screens were undertaken in the past to find compounds that could reverse HD-related changes in cells expressing µHtt, such as the accumulation/aggregation of mutant protein or downstream effects on cellular processes.21,22 For instance, enhanced clearance of overexpressed µHtt served as a readout for a time-resolved fluorescence resonance energy transfer–based high-throughput screen of 1.5 million compounds in a hippocampal cell line expressing a 573–amino acid µHtt fragment that identified several hit structures. 23 A similar assay with PC12 cells and µHtt-EGFP fusion protein also found a novel compound that was selective for the mutated protein and protective against µHtt-induced toxicity. 24 Other screens used a reduction in aggregation of µHtt 25 or inhibition of cell death26–28 as a primary readout. In all cases, only a polyQ-containing N-terminal fragment was expressed in the cells, as compared with the full-length Htt protein, due to its propensity to form aggregates and cytotoxic effects.
Reasoning that correcting aspects of µHtt-mediated transcriptional dysregulation might result in protective effects, we developed a high-throughout screening (HTS) assay for the identification of compounds that could modulate one of the most validated transcriptional effects of µHtt—namely, interference with CREB-mediated transcription. Although earlier studies demonstrated the impact of µHtt expression on transcription using an N-terminal polyQ µHtt fragment and a CRE-controlled reporter gene, 15 we chose to design an assay with the full-length Htt protein containing a polyQ tract with 138 glutamines. In fact, the cAMP pathway is known to be also dysregulated when full-length µHtt is expressed,17,29 and a phenotypic screen in a µHtt full-length model might sample a wider range of mechanisms than those represented in a µHtt fragment model. The characterization of recombinant cell clones, assay performance, and results from the screen are presented. This represents the first report of a phenotypic screen in a novel full-length µHtt cell model that can be employed in further screening campaigns aimed at the identification of novel candidate therapeutics for HD employing a characterized, mechanistically relevant transcriptional readout.
Materials and Methods
Reagents
298/T-Rex cells were purchased from Life Technologies (Paisley, UK) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (#21969-035) containing 10% fetal bovine serum (certified U.S. origin; #16000-044), 1% Glutamax (#35050-038), and 1% penicillin/streptomycin (#15140-122), all purchased from Life Technologies, with addition of 5 µg/µL Blasticidin (#ant-bl-1; InvivoGen, San Diego, CA).
Tetracycline-inducible T-REx vector pcDNA5/TO for cloning of full-length huntingtin was also purchased from Life Technologies (#V103320). FuGENE 6 transfection reagent used for transfection was obtained from Roche Applied Science (Basel, Switzerland).
The construct used for CRE-luciferase overexpression was pcDNA3.1/Zeo (#V860-20; Invitrogen, Carlsbad, CA). Y27632 Rho kinase inhibitor was obtained from TOCRIS (#1254; Bristol, UK). IBMX (#I7018) and forskolin (#F6886), used for phosphodiesterase inhibition and stimulation of cAMP levels, respectively, were purchased from Sigma-Aldrich (St. Louis, MO). Luciferase reporter gene activity was measured with the PerkinElmer SteadyLite Plus kit (#6016759; PerkinElmer, Waltham, MA). Cell proliferation and viability were measured with the PerkinElmer ATPlite Assay System (#6016949). pRL-TK (thymidine kinase) was purchased from Promega Corporation (Madison, WI). The pBluescript-II vector was used to increase the amount of DNA in transfections. The pcDNA3.1 Hygro(–)TA-RL plasmid was obtained by cloning the RL insert from pRL-TK into the pcDNA3.1 Hygro(–) vector (Life Technologies) after replacement of the cytomegalovirus (CMV) promoter with TA.
Cloning of Full-Length Huntingtin Genes in Tetracycline-Inducible Vectors
The full-length human huntingtin complementary DNA (cDNA) containing either 17 (wt) or 138 (mutant) CAG repeats was subcloned in the tetracycline-inducible T-REx vector pCDNA5/TO by standard restriction enzymes (REs) (Thermo Fisher Scientific, Waltham, MA) digestion and ligation (T4 DNA ligase; Thermo Fisher Scientific). The starting clones were kindly provided by Dr. David Rubinsztein (pTre2Hyg:-3xFLAG:HD17Q and pTre2Hyg:3xFLAG:HD1-38Q). In brief, the original Htt clone was digested with the restriction enzyme Mlu1, overhang extremities were filled with Klenow polymerase (Life Technologies), and the cDNA was extracted by Not1 digestion. The mutant Htt cDNAs were inserted by a T4 DNA ligation protocol in the recipient plasmid pCDNA5/TO (Life Technologies), previously digested with EcoRV and Not1 REs, generating the two clones pcDNA5/TO- 3xFLAG:HTT17Q and pcDNA5/TO-3xFLAG-HTT138Q, which contain the full-length Htt cDNAs Q17 and Q138, respectively. The two resulting clones were sequenced for quality control and used for the generation of transgenic cell lines.
Generation of the Cell Lines, Cell Culture, and Clone Selection
The FuGENE 6 transfection reagent was used for transfecting pcDNA5/TO-3xFLAG:HTT17Q or pcDNA5/TO-3xFLAG-HTT138Q and pcDNA3.1/Zeo into 293/T-REx cells to generate two different stable cell lines with
138Q (µHtt) + CRE-luciferase
17Q (wtHtt) + CRE-luciferase
In both cases, the expression of the huntingtin protein is inducible by the addition of doxycycline, and the readout is the CRE-mediated luciferase activity. The clones were identified using dilution and ring cloning methods and selected with 1 mg/mL Hygromycin B and 50 µg/mL Zeocin (both from InvivoGen). The best clones were identified by testing them for luciferase activity decrease and adenosine triphosphate (ATP) cell viability (see protocols below) after 72 h of induction with 1 µg/mL doxycycline.
For routine culture, cells were grown in DMEM supplemented with 10% fetal bovine serum and 5 µg/µL Blasticidin, 0.25 mg/mL Hygromycin B, and 50 µg/mL Zeocin and kept in a 37 °C incubator at 95% air/5% carbon dioxide.
Western Blots
293/T-REx Q17 RA1, 293/T-REx Q138 RL1, and 293/T-REx Q138 RB1-di cells were harvested at different induction times (0, 24, and 72 h) and lysed in RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM EDTA, 1% NP40, 0.5% Triton X-100) in the presence of a cocktail of protease inhibitors (Roche Applied Science) and 5 mM sodium butyrate. Then, 30 µg total lysates was resolved on 3% to 8% Tris-Acetate gels (Life Technologies) with Tris/Tricine/sodium dodecyl sulfate running buffer and transferred overnight onto PVDF membranes. The membranes were stained with Ponceau-S (Sigma-Aldrich) to confirm a uniform transfer of proteins and subsequently blocked using nonfat dry milk (Cell Signaling, Danvers, MA).
The membranes were probed with antibodies against the N-terminal (#H7540; Sigma-Aldrich) and C-terminal (clone HDC8A4, #MCA2051; Serotec, Kidlington, UK) region of Htt and corresponding secondary horseradish peroxidase–conjugated antibodies. The antigen-antibody complex was detected using the ECL prime substrate (#RPN2232; Amersham, Little Chalfont, UK). α-Tubulin (clone DM1A, #T9026; Sigma-Aldrich) was used as a loading control.
RNA Extraction, Reverse Transcription, and RT-qPCR
To examine the expression of the Htt transgene, total RNA was purified from plated cells at different time points (after 24 h, 72 h of induction or not induced), using the RNeasy Plus mini kit (QIAGEN GmbH, Hilden, Germany), following the manufacturer’s protocol. To assess RNA integrity, quality control (integrity of 28S and 18S ribosomal RNAs) was performed by ethidium bromide staining of agarose gels (both from Sigma-Aldrich) and quantified by a Nanodrop 2000 (Thermo Fisher Scientific) spectrophotometer.
For each sample, cDNA was synthesized from 1 µg total RNA, using the QuantiTect Reverse Transcription Kit (QIAGEN) according to the instruction provided with the kit.
Reverse transcriptase quantitative PCRs (RT-qPCRs) were carried out with a CFX96/C1000 thermal-cycler instrument (Bio-Rad, Hercules, CA), using a SYBR green master mix (Bio-Rad) with primers that matched at the 3′ end, between base pairs 9050 and 9110, the Htt coding sequence (NM_002111; Htt-9050-for CCGTGGTGTATAA- GGTGTTTCAGA; Htt-9110-rev GACCCAGTCCCGG- ACCAT), and the flag in the plasmid sequence (Htt-flag-for GACATCGATTACAAGGATGAC; Htt-flag-rev GGCCTT- CATCAGCTTTTCCA). All PCRs were run under the same cycling conditions (95 °C/3 min, 95 °C/30 s, 60 °C/30 s, for 40 cycles). The relative abundance of each transcript was determined by a standard curve generated from 10-fold serial dilutions of cDNA amplified by PCR (run protocol: 94 °C/3 min, 94 °C/30 s, 55 °C/30 s, 72 °C/30 s, for 40 cycles) using Taq polymerase (Life Technologies) and the same primers used in the RT-qPCR. They were purified after ethidium bromide agarose gel with the QIAquick PCR purification kit (QIAGEN) following the manufacturer’s instructions. All RT-qPCR data were normalized to RPL13a (NM_012423, RPL13a-for CCTGGA-GGAGAAGAGGA-AAGAGA; RPL13a-rev TTGAGGACCTCTGTGTATT-TGTCAA; run condition above mentioned).
CRE-Luciferase Reporter Assay
The clones that displayed a significant CRE-luciferase activity ratio or a significant ATP concentration ratio between basal and doxycycline-induced conditions (293/T-REx Q138 RL1 and 293/T-REx Q138 RB1-di) were expanded and tested together with the 293/T-REx Q17 RA1 cell line, which was used as a wild-type control.
Based on preliminary experiments with different cell densities and incubation times, the 293/T-REx Q138 RL1 clone was chosen as the best clone for the development of an assay for compound screening.
Cells were counted with a Neubauer chamber and plated at 10,000 cells/well (100 µL) in poly-D-lysine–coated 96-well plates (CulturPlate-96, white, #6005688; PerkinElmer). DMSO or compounds were applied in a 50-µL volume with a final DMSO concentration of 0.5%. Induction of transcription was obtained by applying 1 µg/mL doxycycline in a 50-µL volume, whereas the noninduced basal expression was obtained with the same volume of normal culture medium. The final assay volume was 200 µL. The primary screening was conducted with compounds at 20 µM under induced conditions (µHtt overexpression) with replicates on two independent plates. For the concentration-response determination, the compounds were tested with a throughput of one compound per plate in 1 µg/mL doxycycline-induced condition and for comparison also in normal medium (not induced condition), with a concentration range from 50 to 0.03 µM. For the negative control, cells were treated with 0.5% DMSO in normal culture media. The positive control was 0.5% DMSO with doxycycline-containing medium at 1 µg/mL. The reference compound was the Y27632 Rho kinase inhibitor used at 20 µM and 0.5% DMSO under the induced condition.
The plates were incubated for 72 h at 37 °C at 95% air/5% carbon dioxide. After the incubation, the media were replaced with 40 µL DMEM without phenol red (#31053-028; Life Technologies) containing 0.5 µM IBMX and 1 µM forskolin. The plates were incubated for an additional 5 h at 37 °C at 95% air/5% carbon dioxide, followed by addition of 40 µL/well of the luciferase SteadyLite Plus (PerkinElmer) substrate. The plates were placed on a plate shaker for 30 min at 37 °C before the luminescence signal was read.
A Mithras LB 940 Reader Mithras plate reader (Berthold, Bad Wildbad, Germany) was used for the measurement of luminescence in the samples, with a counting time of 0.1 s/well.
We validated the single-concentration assay also in a higher density 384-well format with a final assay volume of 80 µL per well. 293/T-RExQ138 RL1 cells were seeded at 4000 cells/well in 40 µL medium in poly-D-lysine–coated 384-well white plates (OptiPlate-384, #6007299; PerkinElmer). Then, 20 µL of compound samples was added in a final concentration of 0.5% DMSO, and subsequently 20 µL of 1 µg/mL doxycycline was added. Following a 72-h incubation in a 5% carbon dioxide incubator, the medium was removed and replaced with 25 µL medium without phenol red containing 0.5 µM IBMX and 1 µM forskolin. After 5 h of additional incubation in a 5% carbon dioxide incubator, the luciferase activity was measured by adding 25 µL of the SteadyLite Plus substrate. Luminescence was read after 30 min of shaking at 37 °C with a 0.5-s/well counting time. As reference compound, an internal active molecule (compound D) identified during the screening campaign was used at a concentration of 17 µM.
The validation of the assay was conducted according to the “Assay Guidance Manual.” 30 A 2-day plate uniformity study with 3 plates per day, with an interleaved-signal format alternating high, medium, and low signals, was performed. The high signal was the noninduced DMSO control, the low signal was the doxycycline-induced DMSO control, and the medium signal was obtained with the internal reference compound D.
Data Analysis
For evaluating the induction window, the Z′ and signal window (SW) were calculated using the negative noninduced DMSO control and the positive-induced DMSO control. 31 The normalization applied to each plate was
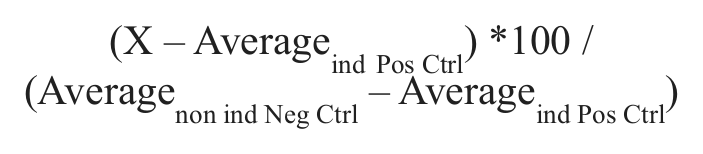
The activity of compounds in a single concentration was evaluated with a threshold of 2 times the standard deviation over the mean of the induced DMSO control.
Concentration-response curves were fitted with the four-parameter logistic nonlinear regression model (model 205) using the IDBS XLfit software (IDBS, Guildford, UK). The activity of compounds in concentration response was normalized independently in noninduced and doxycycline-induced conditions (respective DMSO control = 1).
Transient Transfections with Reporter Constructs
To demonstrate that CRE-luciferase levels were specifically influenced by µHtt expression, we transiently transfected the 293/T-REx Q138 RL1 with a Renilla luciferase reporter gene under the control of TA or TK promoters. Ten thousand cells per well were seeded in a 96-well plate without antibiotics. After 24 h, the cells were transfected with Lipofectamine 2000 2.5× (Life Technologies) and TA-Renilla or TK-Renilla plus control construct pBlueScript, or pBlueScript alone, for a total amount of 0.2 µg DNA per well.
The medium was replaced with fresh culture medium after 3.5 h, and after 24 h, 1 µg/mL doxycycline was applied to the wells. After 2 days, cells were lysed, and firefly and Renilla luciferase levels were read on a Berthold Mithras LB940 using the Dual-Luciferase Reporter assay system from Promega, according to the manufacturer’s instructions.
ATP Assay
For the additional characterization of cell clones or hit compounds, a cell viability assay measuring ATP levels was established. Cells were counted with a Neubauer chamber, and 5000 cells/100 µL DMEM medium with 10% FBS were transferred into each well of a poly-D-lysine–coated 96-well white plate.
The treatment of cells with compounds followed the same protocol as described in the “CRE-Luciferase Reporter Assay” section. After 72 h of incubation, the medium was removed, and 100 µL DMEM without phenol red was added. Total ATP levels reflecting cell number and proliferation were determined using the PerkinElmer ATPlite Assay System as recommended from the manufacturer. The Mithras plate reader (Berthold) was used for measurement of luminescence intensity with a counting time of 0.1 s/well.
INCUCYTE Kinetic Imaging System
Further analysis of cell clones and effects of hit compounds was carried out with the INCUCYTE (Essen Bioscience, Ann Arbor, MI) phase-contrast microscope that was placed inside a standard tissue culture incubator.
An experiment with the same design used for the ATP viability test was set up. 293/T-REx Q138 RB1-di cells were seeded in a 96-well plate and treated with 20 µM of the Y27632 control compound or with hits that were identified during the screening campaign as well as with 1 µg/mL doxycycline. Induced and noninduced DMSO controls were added.
The plate was incubated for 72 h at 37 °C and 95% air/5% carbon dioxide under control of the INCUCYTE instrument, which acquired one image every 3 h.
Selection of Compound Library
The screening compound collection was made up of about 24,000 unique compounds. This set was mainly selected from our corporate library, which consisted of about 90,000 molecules. Approximately 80% of this library has been cherry-picked from commercial sources to cover a wide structural diversity while sampling suitable drug-like physicochemical properties. Together with the diversity set (>90% of compounds), some small focused sets were also selected, being represented by aspartyl protease inhibitors, calpain inhibitors, and aggregation inhibitors, among others. Confirmed primary actives were checked by preliminary quality control (pQC: liquid chromatography/mass spectrometry [LC/MS] analysis, compound integrity >80%). The activity seen in the primary screen was confirmed with a different batch of the compound that was characterized by a full quality control analysis (fQC: nuclear magnetic resonance [NMR], LC/MS, compound integrity >90%, structure confirmation), tested in a concentration-response assay followed by a compound specificity evaluation.
Results
Assay Principle
We wanted to establish a phenotypic primary screening model in which the pathogenic transcriptional dysregulation, driven by expression of mutant huntingtin, is used as a readout for the identification of small molecules that may slow, halt, or reverse disease progression.
To that end, we generated two 293/T-REx recombinant cell lines containing tetracycline-inducible forms of the wild-type (Htt) or the mutant full-length human huntingtin protein (µHtt), bearing either 17 (Q17) or 138 (Q138) polyglutamine repeats at the N-terminus. To monitor the effect of Htt expression on gene transcription, a reporter gene construct in which luciferase expression is controlled by CRE sequences in the promoter (CRE-Luciferase construct) was co-transfected in the same cell lines. µHtt is known to impair the cAMP-dependent transcription of genes controlled by the CRE promoter by sequestering the CBP transcription factor ( Fig. 1 ). In our model, the presence of µHtt leads to a decrease of forskolin-induced luciferase protein synthesis. Among several isolated clones, three were selected and characterized—namely, the 293/T-REx Q17-RA1 clone, expressing the wild-type form of Htt, and the 293/T-Rex Q138 RL1 and Q138 RB1-di clones, both expressing the mutant form of the protein.
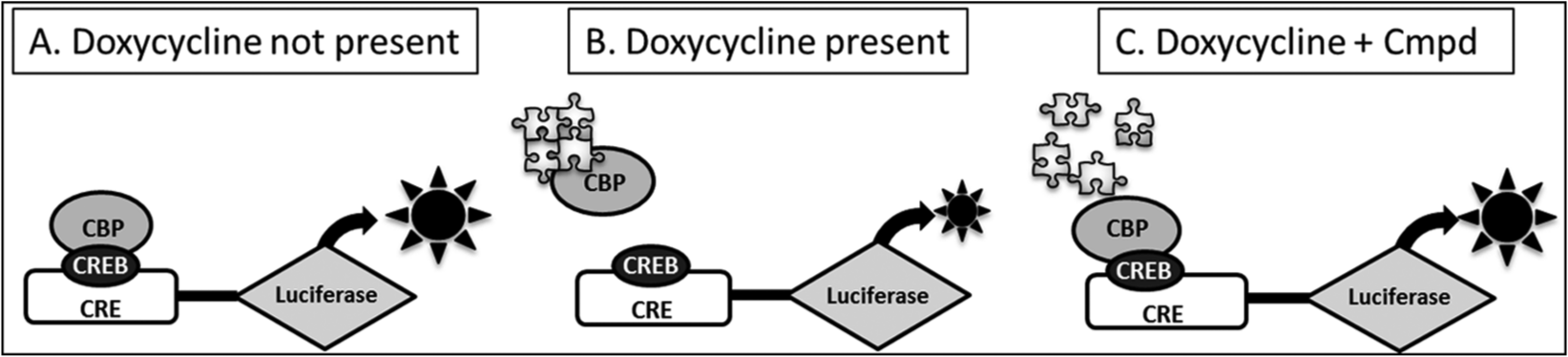
Principle of cAMP responsive element (CRE) luciferase screening assay. The 293/T-REx recombinant cell lines express the full-length mutated Htt protein (µHtt) with 138 glutamine repeats under control of the inducible Tet operon and a luciferase reporter gene under control of the CRE. The CRE signaling pathway can be induced by addition of forskolin that activates the cAMP-generating enzyme adenylyl cyclase. In the presence of cAMP, the transcription factor CRE binding protein (CREB) binds to the CRE and to the CREB binding protein (CBP), which leads to transcription of the luciferase reporter gene. (
Clone Characterization and Development of Reporter Screening Assay
The expression of the full-length Htt protein was evaluated by Western blot in the recombinant T-REx clones using antibodies against N-terminal (3–16aa) and C-terminal (2703–2911aa) epitopes. A clear band at the predicted molecular weight of 347 kDa for full-length Q17 Htt and of 363 kDa for full-length Q138 Htt (reference sequence taken from RefSeq NCBI Database NP_002102.4) was detected in all the clones, confirming the doxycycline induction response. N-terminal fragments of Htt were also detected for the Q138 RL1 clone ( Fig. 2A ).
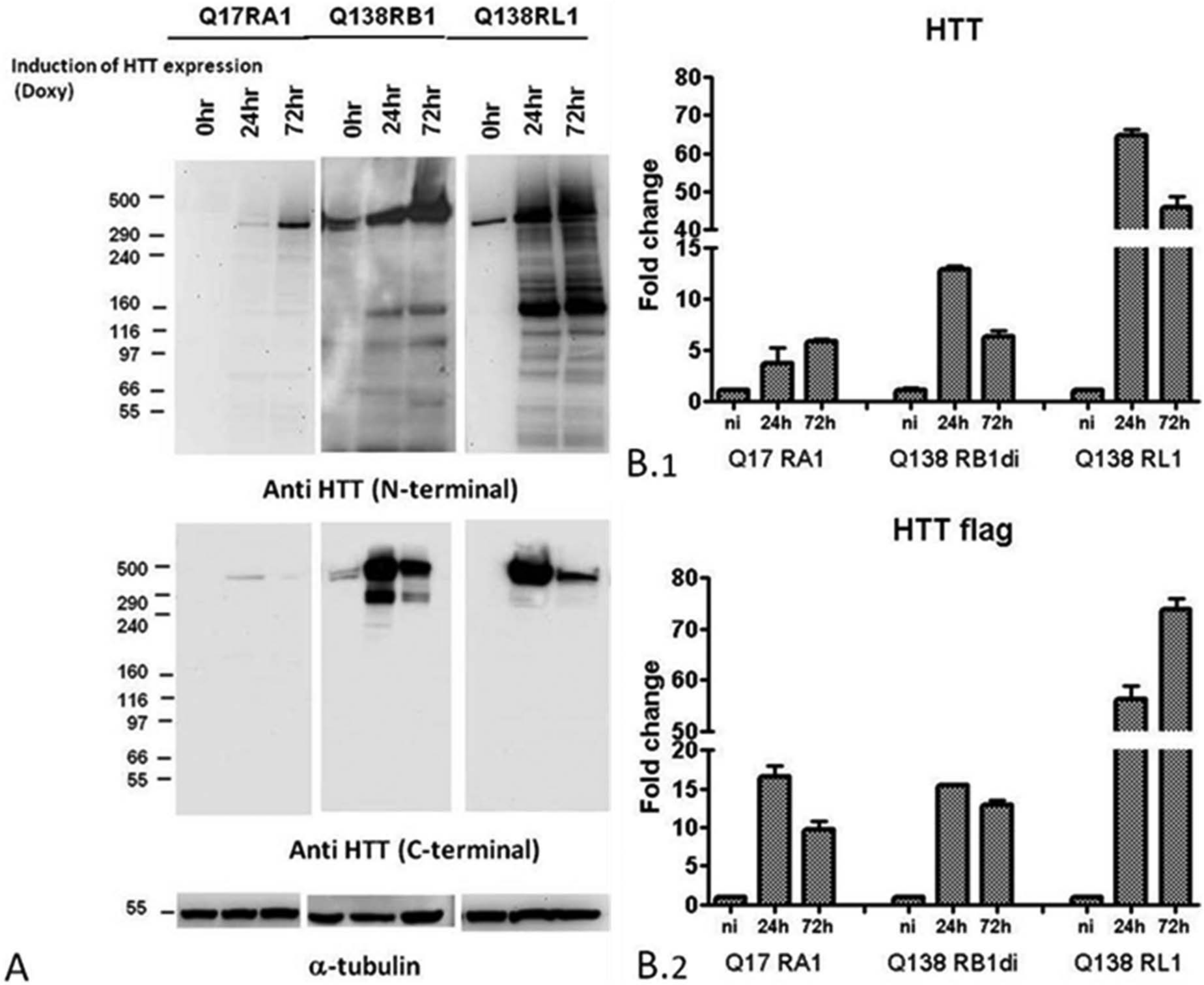
Characterization of huntingtin-expressing cell lines. (
Transgene expression in the recombinant clones was evaluated by RT-qPCR with two different primer sets, matched at 5′ and 3′ ends, that confirmed the presence of the inducible system and assessed an increase of Htt messenger RNA (mRNA) levels in doxycycline-treated clones with respect to untreated cells ( Fig. 2B ). The transgene expression was investigated at the two different time points of 24 h and 72 h after doxycycline induction. Results confirmed the inducibility of transgene mRNA steady state in the different clones following doxycycline addition, with fold increases over baseline that were comparable for clones Q17 RA1 and Q138 RB1.
For the development of the phenotypic assay, the three clones were induced for 72 h with doxycycline and analyzed for luciferase activity (reporter assay) and cell viability (ATP assay) ( Fig. 3A , B , respectively). As expected, the 293/T-REx Q17 RA1 clone showed no effect on transcription and no cellular toxicity upon doxycycline induction. On the contrary, Q138 RB1-di and Q138 RL1 cells displayed a strong reduction of CRE-luciferase activity ( Fig. 3A ). The ratio between luciferase signals of noninduced and doxycycline-induced cells was higher for the 293/T-REx Q138 RL1 clone than for 293/T-REx Q138 RB1-di clone. A reference compound Y27632, previously demonstrated to reduce µHtt effects in various HD preclinical models, 32 was tested in 293/T-REx Q138 RL1 cells. This compound was able to partially restore the CRE-mediated transcription observed when µHtt expression was induced with doxycycline conditions ( Fig. 3C ). In the viability assay, the 293/T-REx Q138 RL1 clone did not show any cytostatic effect in induced conditions, as was observed for the 293/T-REx Q138 RB1-di clone ( Fig. 3B ). With the latter clone, the Y27632 reference compound was able to partially restore cell proliferation under induced conditions, again with a smaller effect also in noninduced conditions ( Fig. 3D ). The activity observed under uninduced conditions may be due to the modulation of effects mediated by leaky transgene expression (see Fig. 2A ).
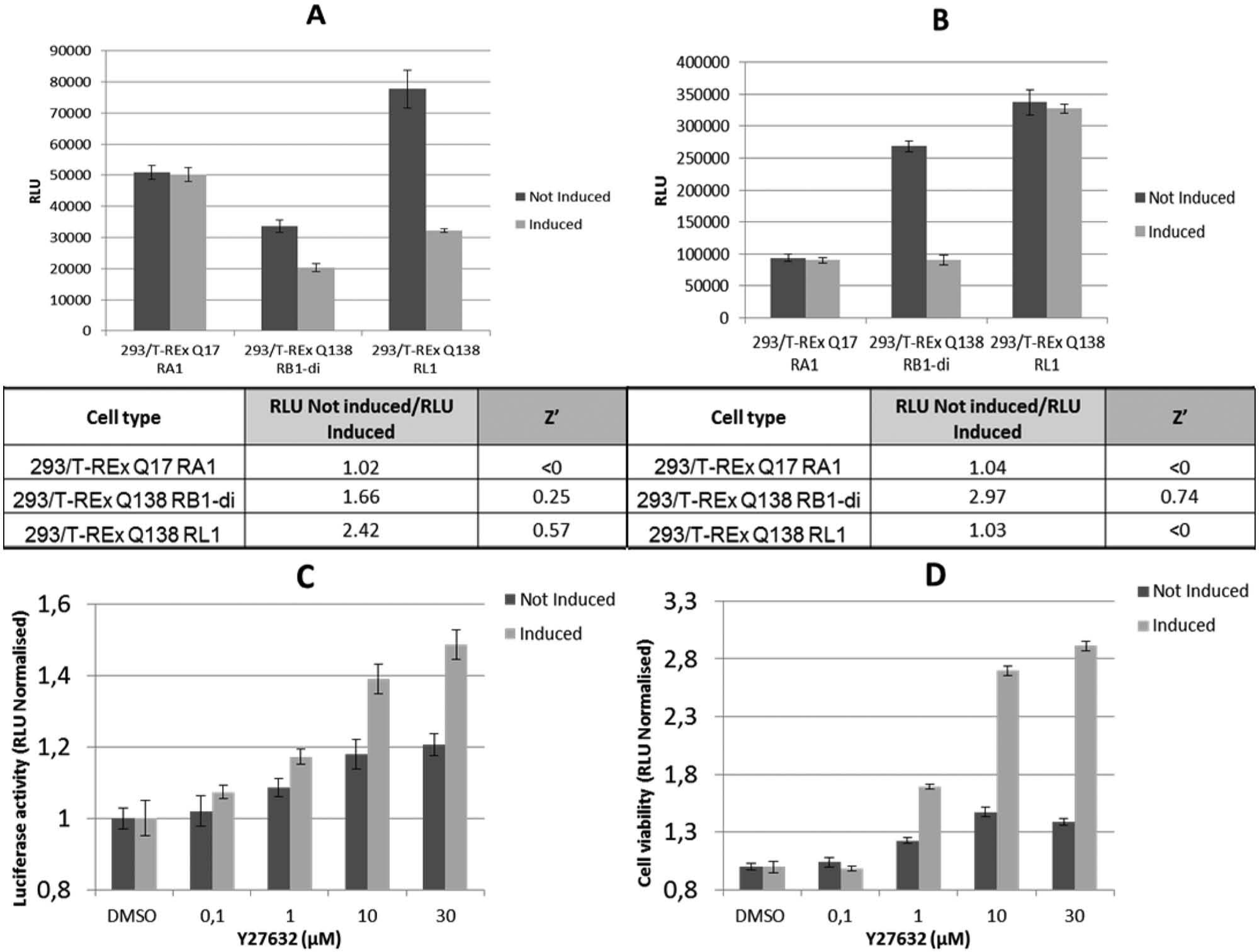
(
To confirm the cytostatic effect of µHtt in the Q138 RB1-di clone, we performed an experiment with the INCUCYTE Live-Cell Imaging System in which cell proliferation was measured in real time. The images acquired by the instrument show a significant decrease in cell density between the basal condition and the condition after 72 h of induction with doxycycline (
The larger noninduced to doxycycline-induced signal ratio in the reporter assay, as well as the absence of a cytostatic effect caused by Htt in the Q138 RL1 clone, was in our opinion a clear advantage for the development of a phenotypic assay that would be sensitive to weak compounds. For that reason, we focused all further reporter assay optimization efforts for the HTS assay on the Q138 RL1 cell clone.
Effect of Huntingtin Expression on Reporter Gene Systems
To demonstrate that the modulation of CRE promoter activity by mutant huntingtin upon doxycycline treatment was a specific effect on CREB-mediated transcription, we transiently transfected the 293/T-REx Q138 RL1 clone with Renilla luciferase under the control of TA or TK control promoters and measured the effects of transgene induction on these reporters. The experiments were conducted in 96-well plates with doxycycline induction for 48 h. The dual luciferase measurement showed, as expected, a clear decrease of firefly luciferase (from the CRE-regulated reporter) levels when mutant huntingtin expression is induced, with an average ratio between noninduced and induced cells varying between 1.56 and 1.78 (
Reporter Assay Optimization for HTS in a 96-Well Format
For the optimization of the assay conditions with the 293/T-REx Q138 RL1 clone, different incubation times with doxy-cycline were tested (
Primary Screening Results
The developed 96-well reporter gene assay was used in an automated format to screen a diverse library of 24,000 compounds at a concentration of 20 µM. As a positive control, the previously described Rho kinase inhibitor (Y27362) was employed.
The postscreening analysis of assay robustness and reproducibility showed low variability and high separation of induced and noninduced controls. The Z′ and SW from a representative subset of screening plates were 0.57 and 6.7, respectively (
Hit Confirmation in Concentration Response
The primary hits were tested for concentration response in induced and noninduced conditions on the same plate, thus allowing a simultaneous assessment of activity (on induced cells) and specificity (response on induced cells as compared with noninduced cells).
We expected that for some of the compounds, the effect seen on luciferase activity in the induced condition might be due to an unspecific effect on the luciferase itself or on transcription itself. This was already seen for the Y27632 reference compound, whose unspecific increase of luciferase activity is probably due to a cytoprotective and pro-proliferative effect of the compound33,34 or to specific compound effects on transgene-derived µHTT protein under basal conditions, as the inducible system shows a degree of leakiness ( Fig. 2 ). To profile the active compounds in terms of specificity of the observed readout, we compared the maximum efficacy calculated from the concentration-response curve fit of the normalized luciferase activity of the µHtt-expressing cells (Emax ind) and of the noninduced cells (Emax non ind) and set the following criteria: (1) Compounds showing a difference between Emax non ind and Emax ind below 0.3 were considered nonspecific ( Fig. 4A ), (2) compounds active both on induced and noninduced cells and showing a difference between Emax non ind and Emax ind higher than or equal to 0.3 were considered partially specific ( Fig. 4B ), and (3) compounds active on induced cells and showing no significant activity on noninduced cells at any of the tested concentrations were considered specific ( Fig. 4C ).
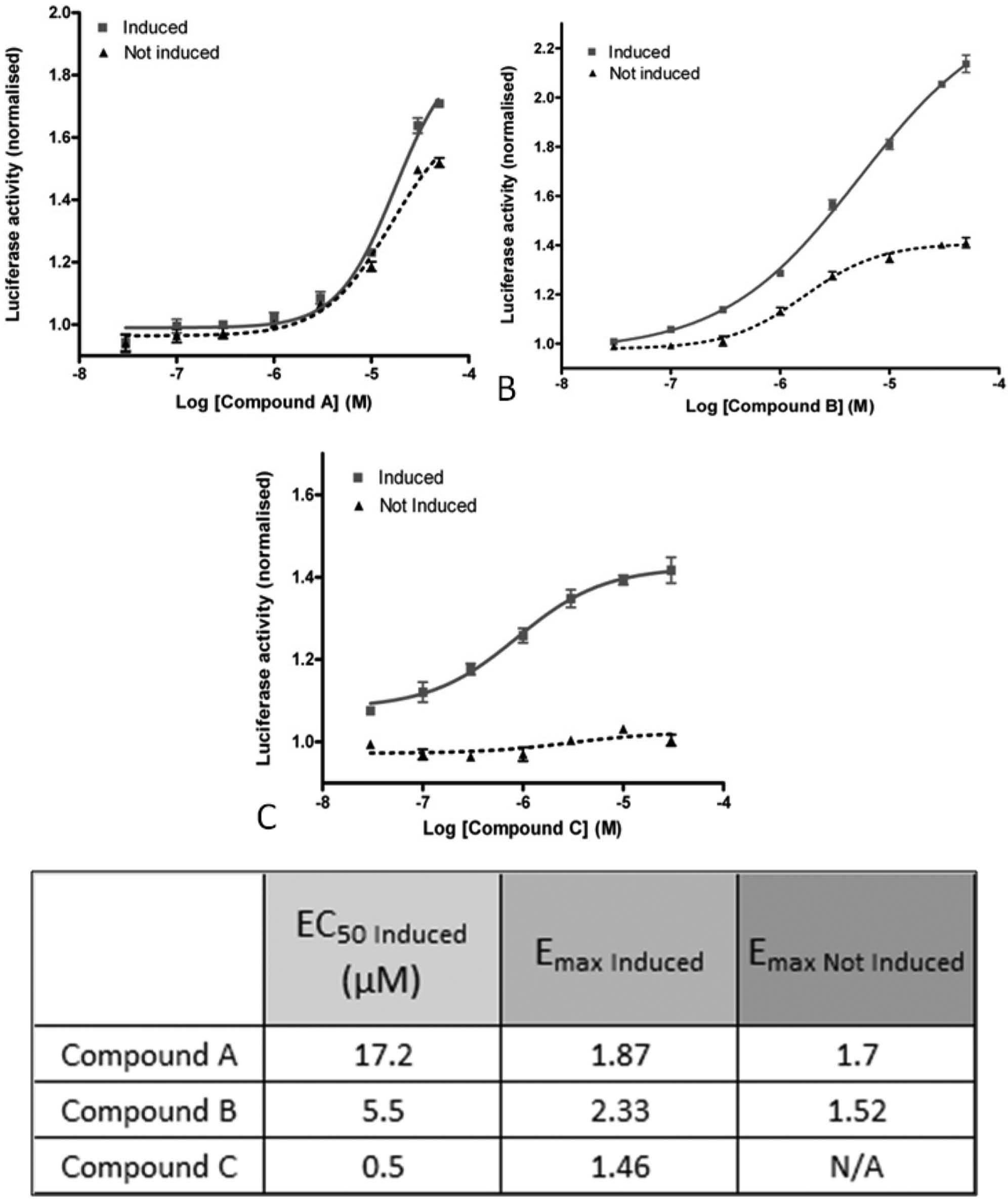
Concentration-response curves of active compounds in the reporter gene luciferase assay in the 96-well plate. The figure shows examples of three different compound behaviors. Doxycycline-induced and noninduced 293/T-REx Q138 RL1 cells were incubated with compounds at eight concentrations (from 50–0.03 µM) for 72 h. The classification was made based on the difference between the maximum efficacy calculated from the concentration-response curve fit of normalized luciferase activity in the mutant huntingtin-expressing cells (Emax Ind ) and in the noninduced cells (Emax Non Ind). (
The compound potency was estimated from the EC50 on the induced cells.
Analysis of Hit Compounds
The 1188 active compounds from the single concentration screen were first analyzed with our pQC protocol. This resulted in 1.091 confirmed actives (compounds with confirmed activity at single concentration, pQC passed).
This set of compounds underwent a clusterization, using the Tanimoto distance for the structure similarity assessment. A prioritization was made, and representative compounds of the most interesting clusters were repurchased. Of 228 compounds tested as a new batch in EC50 and fQC, the activity was confirmed for 186 compounds, whereas the remaining 42 resulted in either not being active or in a failed fQC. Of this set of 186 active compounds, only 47 could be classified as specific, yielding a final hit rate of 0.19% for the screening campaign. This final set of compounds was subgrouped into 10 series by using the original cluster analysis and a visual inspection for structural similarity. Each series was populated by at least two compounds, whereas the rest of the molecules were classified as singletons. Subsequently, these initial series were expanded with close analogues selected from commercial sources, providing the final series composition as reported in Table 1 .
Summary of Hit Compounds.
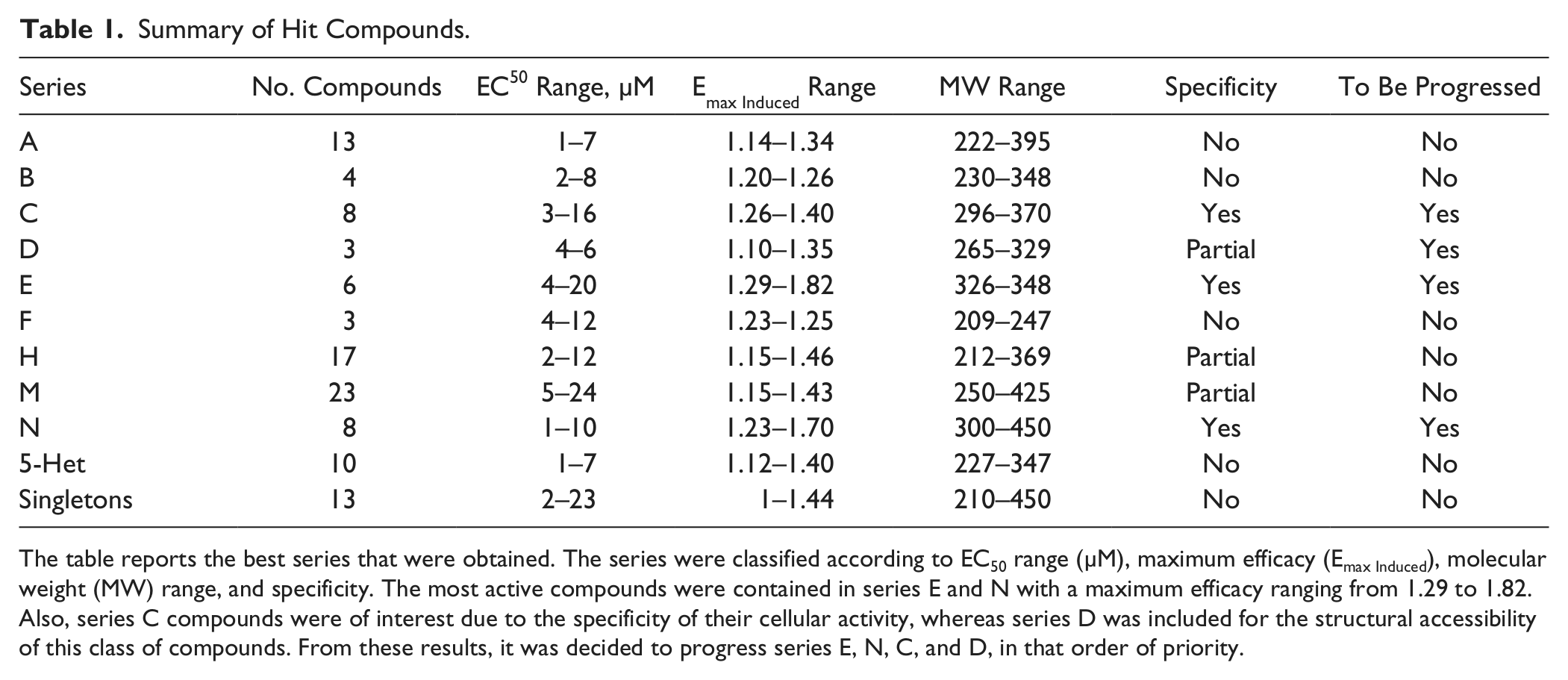
The table reports the best series that were obtained. The series were classified according to EC50 range (µM), maximum efficacy (Emax Induced), molecular weight (MW) range, and specificity. The most active compounds were contained in series E and N with a maximum efficacy ranging from 1.29 to 1.82. Also, series C compounds were of interest due to the specificity of their cellular activity, whereas series D was included for the structural accessibility of this class of compounds. From these results, it was decided to progress series E, N, C, and D, in that order of priority.
Table 1 shows the EC50 and Emax Induced range for each series. Each of the series was analyzed and evaluated also in terms of chemical feasibility, analogue expansion, IP position, and specificity against the noninduced cell line. The most active compounds were obtained from series E and N, with a maximum efficacy ranging from 1.29 to 1.82 and an optimal preliminary ADME profile (data not shown). Series C compounds were interesting for their specificity, whereas series D was included based on the synthetic accessibility of this class of compounds. Those compound series for which specificity could not be demonstrated throughout were disregarded. Finally, series E, N, C, and D were prioritized and progressed into a hit-to-lead phase.
Assay Adaptation to a 384-Well Format
To enable future use of the assay for screening larger compound libraries, we miniaturized the assay to a 384-well format. The positive control used was compound D, identified during the primary screening campaign in 96-well plates. The compound showed an EC50 of 6 µM and a maximum increase of the induced signal of 1.85 when normalized to the induced DMSO control (data not shown). The compound was tested at a concentration that resulted in about half of the recovery of the luciferase transcription dysregulation caused by the doxycycline-induced expression of mutant Htt. The concentration was calculated as 17 µM from the induced concentration-response curve (not shown). The same concentration was applied to the medium signal columns in the plate uniformity test in order to have the estimation of the recovery of 50% of luciferase activity. The plate uniformity test was executed with three plates of alternating columns of high (H), low (L), and medium (M) signals, repeated in an independent experiment over 2 days. This study confirmed that the assay could be transferred to 384-well plates (
Discussion
In the present work, we describe the development of an assay that is able to model an early event in the pathogenesis of HD through the expression of full-length mutated huntingtin and the associated downregulation of the CRE signaling pathway. The choice of the full-length form of Htt, rather than exon-1 or other Htt fragments that were reported previously,24–27 enabled a phenotypic screen using a much more disease-relevant cellular phenotype that included the proteolytic processing of Htt and possibly other pathological mechanisms that are not strictly related to the N-terminus of the protein.
293/HEK cells, instead of neural cell lines, were selected as cell background because of their suitability to large-scale screening activities, especially in miniaturized assay formats as employed in HTS campaigns. Moreover, HEK cells have been shown to display a number of molecular signatures typical of neuronal lineage cell lines. 35 The presence of mRNA and gene products typically found in neurons; neurofilament (NF) subunits NF-L, NF-M, and NF-H; and α-internexin, among others, reveals neuronal progenitor properties of HEK cells. In our assay, HEK cells clearly showed the expected deficits after Htt expression, as observed in “more relevant” cell systems.
We generated stable cell lines expressing both the CRE-luciferase reporter gene and either the wild-type Q17 or mutated Q138 form of the full-length huntingtin gene under the control of a doxycycline-inducible CMV promoter. Transcription dysregulation was observed only for cells expressing Q138 µHtt, whose concentration might have surpassed a threshold required for the initiation of a cascade of toxic events that ultimately led to an antiproliferative effect. The µHtt-mediated effect on transcription was specific for the CRE promoter since no alteration in reporter gene expression was observed when different promoters were used.
Different cell clones showed a different sensitivity to the impairment of transcription and cell proliferation, which could depend on different integration sites and result in differences in the expression levels of huntingtin. Starting from a selected pool, we characterized the two best clones for the reporter gene activity and the cell viability readouts that were then optimized and validated as primary and secondary screening assays, respectively. The 293/T-REx Q138 RL1 clone showed a greater noninduced to doxycycline-induced signal ratio in the reporter assay and an absence of a cytostatic effect caused by the mutant Htt expression. These cells provide a phenotypic model that is sensitive to compounds that are too weak to prevent cell damage but are still valuable starting points for medicinal chemistry efforts. 16
The Rock inhibitor Y27632 was used as a tool compound for the initial proof of concept of these assays, exploiting its known antiaggregation properties.33,34 Besides a significant reversal of the inhibition of luciferase activity in induced conditions, a still significant but lower effect could be seen in noninduced cells. In our opinion, this effect might be due to the generic cytoprotective role of Y27632, which was already noted for human embryonic cells and other cell lines and was also detected in an ATPlite assay with 293/T-REx Q138 CRE RB1-di cells. In addition, the RB1 clone displays a degree of transgene expression leakage in the absence of doxycycline. Nevertheless, the significant difference, observed already at a concentration of 1 µM, indicates a clear protection from µHtt-induced impairment of transcription and cell proliferation.
The partial specificity that was demonstrated by the Rock inhibitor Y27632 was the hit threshold that we aimed to surpass with our screening campaign. The criteria for the classification of the compounds included therefore the threshold of 0.3 that was the average of the observed difference in Emax between the induced and noninduced condition for the Y27632 compound.
The assay is amenable to HTS applications as demonstrated by its validation with a 384-well format, showing a high reproducibility and robustness, although we did not have the need to screen our limited number of compounds in that format.
Using the assay described above, we screened a library of about 24,000 diverse compounds and identified several hit series, each of these showing a different level of specificity on CRE-mediated transcriptional regulation. The series identified are currently being optimized and tested in different disease-relevant secondary assays to investigate their mode of action, particularly their effect on mutant huntingtin aggregation assays. We believe that the assay described here represents a valuable in vitro model for the discovery of novel hit series that, once confirmed in other relevant secondary assays, could represent good candidates for the development of drugs that are able to modify disease progression in Huntington’s disease.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.