
Abstract
A persistent problem in early small-molecule drug discovery is the frequent lack of rank-order correlation between biochemical potencies derived from initial screens using purified proteins and the diminished potency and efficacy observed in subsequent disease-relevant cellular phenotypic assays. The introduction of the cellular thermal shift assay (CETSA) has bridged this gap by enabling assessment of drug target engagement directly in live cells based on ligand-induced changes in protein thermal stability. Initial success in applying CETSA across multiple drug target classes motivated our investigation into replacing the low-throughput, manually intensive Western blot readout with a quantitative, automated higher-throughput assay that would provide sufficient capacity to use CETSA as a primary hit qualification strategy. We introduce a high-throughput dose-response cellular thermal shift assay (HTDR-CETSA), a single-pot homogenous assay adapted for high-density microtiter plate format. The assay features titratable BacMam expression of full-length target proteins fused to the DiscoverX 42 amino acid ePL tag in HeLa suspension cells, facilitating enzyme fragment complementation–based chemiluminescent quantification of ligand-stabilized soluble protein. This simplified format can accommodate determination of full-dose CETSA curves for hundreds of individual compounds/analyst/day in replicates. HTDR-CETSA data generated for substrate site and alternate binding mode inhibitors of the histone-lysine N-methyltransferase SMYD3 in HeLa suspension cells demonstrate excellent correlation with rank-order potencies observed in cellular mechanistic assays and direct translation to target engagement of endogenous Smyd3 in cancer-relevant cell lines. We envision this workflow to be generically applicable to HTDR-CETSA screening spanning a wide variety of soluble intracellular protein target classes.
Keywords
Introduction
Among the many formidable challenges in drug discovery and development, lack of efficacy in phase 2 clinical studies has been cited as the primary driver of drug attrition among major pharmaceutical companies. Retrospective analyses of late-stage trials suggest that key determinants for improving drug discovery processes that have the highest likelihood of translating to clinical success are demonstration of (1) exposure of the drug at the target site of action, (2) engagement of the drug with its molecular target, and (3) a functional pharmacologic activity leading to desired phenotypic outcomes.1,2
Historically, demonstration of small-molecule engagement with its protein target in living systems has been difficult to measure; instead, it has been inferred either indirectly through proximal functional readouts such as monitoring an enzyme-substrate product linked with downstream cellular responses or by use of modified ligands and/or targets. The recent introduction of the cellular thermal shift assay (CETSA) has greatly facilitated the ability to directly monitor drug binding to target proteins in cells and tissue samples.3,4 Traditional thermal shift assays are based on the biophysical principle that thermally induced unfolding of a protein produces a distinct melting curve from which a characteristic melting temperature (Tm) can be extrapolated; these melting curves are often shifted to a higher temperature when a bound ligand stabilizes the protein, evidenced as a thermal shift (ΔTm). Thermal shift assays are well established and highly embedded in drug discovery using purified proteins.5,6 The discovery that this observation of ligand-induced thermal stabilization can also be extended to living systems resulted in the development of CETSA technology, which has for the first time facilitated the routine study of drug binding to target proteins in their relevant cellular context.3,4,7,8
A particularly attractive feature of CETSA is the ability to use unmodified drug ligands to evaluate target engagement directly in disease-relevant cell types; this presents an opportune assay system in which the native cellular protein target is presented at endogenous expression levels and contains the appropriate repertoire of dynamic posttranslational modifications allowing for proper subcelullar localization and recruitment to multimember protein complexes driving disease biology. The high physiological relevance of this system cannot be readily recapitulated in vitro using isolated recombinant protein. In addition, the simplicity of the original CETSA method presents a very low barrier to entry; besides the requirements of a drug candidate and a cell line expressing the protein target, no specialized reagents are necessary beyond a single antibody reagent for quantification of remaining soluble target protein by Western blot analysis.
Initial exploratory evaluations conducted at GlaxoSmithKline using the original CETSA methodology provided compelling data indicating the technique was robust and reproducible in whole cells and lysates, for both CETSA temperature gradient and isothermal dose-response fingerprint (ITDRF) measurements. Confirmation of target engagement using tool inhibitors and lead optimization intermediates enabled decision making for internal drug discovery programs against diverse target classes including protein deactylase, serine/threonine/tyrosine protein kinase, lysine and arginine N-methyltransferases, oxidoreductase, dioxygenase, and E3 substrate adapter protein family members. Our enthusiasm of the broad applicability of CETSA was buoyed by numerous early reports using the technique to confirm target engagement of individual proteins comprising a wide range of target families9–22 and the internal development of a mass spectrometry–based platform for thermal proteome profiling of the entire melting proteome.23,24
We recognized that by replacing the low-throughput, manually intensive Western blot readout originally described with a quantitative, automated, high-density microplate format, we could provide sufficient assay capacity to fully leverage the power of CETSA and embed the technique as an integral part of hit identification and lead generation in early drug discovery campaigns. Such an assay would be pivotal in reconciling the frequent disconnect and lack of rank-order correlation between enzyme Ki values determined from biochemical screens and the corresponding EC50 for the cellular effect, or “cell drop-off.” 25 Rank ordering of compound cellular potency based on measuring quantitative EC50 values for target engagement would facilitate early triage during hit identification campaigns, allowing chemists to focus primarily on structural series possessing desirable cellular activity as a starting point for series expansion. During the lead generation and optimization phase, integration of data providing a quantitative assessment of cellular target engagement, rather than sole reliance on the initial biochemical in vitro data, would greatly inform structure-activity relationships, translating to meaningful improvements in cellular potency. In addition, information provided by CETSA could complement or potentially replace a cellular mechanistic assay and provide a fast track to test only the most promising candidates in expensive and often difficult-to-obtain human disease-relevant primary cells and animal models. We predict that the ability to measure and gain a detailed understanding of engagement of a drug with its molecular target at the earliest stages of drug discovery is expected to be a driving factor to reduce attrition and improve success in the clinic.
In this report, we describe development and application of a high-throughput dose-response cellular thermal shift assay (HTDR-CETSA), a streamlined and miniaturized single-pot homogenous assay adapted for high-density 384-well microtiter plate format. The evolutionary reconfiguration of the original CETSAITDRF procedure for high-throughput applications required addressing multiple challenges: (1) simplification of compound treatment, transient heat denaturation, and lysis regimes permitting elimination of all wash and centrifugation steps with incorporation of requisite automation and (2) introduction of a detection system compatible with a homogenous readout, which would facilitate selective quantification of remaining ligand-stabilized soluble target protein in the presence of both denatured and aggregated target protein, as well as protein lysate and cellular debris.
A key requirement predicating choice of detection modality was that it was generically and facilely applicable, thereby significantly shortening assay development time by eliminating the lengthy task of evaluating and validating suitable target-specific antibody pairs for each new assay. Importantly, it was critical that introduction of the reporter construct neither alter the Tm of the tagged target protein relative to the endogenous cellular counterpart nor interfere with the magnitude of thermal stabilization induced by interaction with a drug molecule. We found that the enzyme fragment complementation (EFC) technology developed by DiscoverX (Fremont, CA) was able to fit these aspirational criteria. 26 HTDR-CETSA features adaptation of EFC as a detection platform, accomplished through expression of full-length target proteins fused to the DiscoverX 42-amino acid ePL tag. Upon addition of the enzyme acceptor (EA) and substrate, the subunits associate to form a fully competent β-galactosidase enzyme, and a chemiluminescent signal is produced. 26 HTDR-CETSA profiling of 123 substrate and alternative binding mode inhibitors of histone-lysine N-methyltransferase SMYD3 indicates the assay platform will provide target engagement data highly predictive of cellular activity in disease-relevant cells.
Materials and Methods
Reagents
Anti-Smyd3 (ab183498 rabbit monoclonal) and anti-IDO1 (ab76157 rabbit monoclonal) antibodies were purchased from Abcam (Cambridge, MA). A monoclonal mouse anti-ePL antibody was generated internally. The IR-800 conjugated goat anti-mouse IgG and goat anti-rabbit IgG were obtained from Millipore (Billerica, MA). The human interferon γ (IFNγ) used for induction of endogenous IDO1 expression was from Sigma-Aldrich (St. Louis, MO). The InCELL Pulse Detection Kit from DiscoverX was used for ePL detection. SMYD3 In-Cell Western assays used antibody reagents for anti-Smyd3 (Santa Cruz Biotechnology, Santa Cruz, CA; sc-67210), anti-HA (Covance, Princeton, NJ: MSS-10 IR HA.11, 16B12), and rabbit anti-MEKK2-K260me (YenZym, South San Francisco, CA; custom). Modified RIPA buffer (#20-188) was purchased from EMD Millipore (Darmstadt, Germany).
Cell Lines
The SC-OV-3 cell line was purchased from ATCC (Manassas, VA; HTB-77, depositors G Trempe and LJ Old) with permission from Memorial Sloan-Kettering Cancer Center and grown under recommended conditions. The MDA-MB-453 cell line was purchased from ATCC (HTB-131, depositor R Cailleau) with permission from The University of Texas M.D. Anderson Cancer Center and grown under recommended conditions. The U-2 OS cell line was purchased from ATCC (HTB-96, depositor Hellstrom) and grown under recommended conditions. The A549 cell line was purchased from ATCC (CCL-185, depositor M Lieber) and grown under recommended conditions.
Constructs and Virus Generation
Full-length human indoleamine 2,3-dioxygenase 1 (IDO1) sequence consistent with the reference sequence NM_002164/GeneID #3620 with a 10 amino acids spacer and ePL fused to its carboxy terminal end and the full-length human Smyd3 sequence consistent with the reference sequence of NM_001167740/GeneID #64754 with ePL directly fused to its carboxy terminal end were chemically synthesized and then cloned into a modified version of the pHTBV1 BacMam expression vector. 27 Recombinant BacMam viruses were generated as described in the Bac-to-Bac kit instructions (Invitrogen, Carlsbad, CA).
Cell Culture
The HeLa S3 cell line (CCL-2.2, ATCC) was adapted for suspension growth. 28 Cells were cultured in DMEM/F12 (1:1 ratio) medium with 4.5 g/L glucose, 2 mM glutamine supplemented with 10% fetal bovine serum. The cell cultures were maintained at 37 °C in a humidified 5% CO2 atmosphere and with gentle shaking at 200 rpm to keep the cells suspended. For BacMam transduction, virus was added to the cells at an approximate cell density of 2e6/mL (see text for multiplicity of infection [MOI], varied depending on experiment). Twenty-four hours posttransduction, culture medium was replaced with low serum DMEM/F12 containing 1% fetal bovine serum (FBS) and treated with 10 µM tool compound or DMSO as a control, sealed with adhesive aluminum foil, and agitated for 2 h shaking at 1200 rpm at 37 °C on a plate Thermo-Shaker PST-100HL (Biosan Laboratories, Warren, MI). The treated cells were analyzed for protein expression and thermal profiling by either Western or EFC ePL readout.
Compounds
[3-(4-(diisobutylamino)-3-((3-trifluoromethyl)-1,2,4-thiadiazol-5-yl)amino)phenyl)butanoic acid], which was termed GSK5628, was synthesized by the HIV Discovery Performance Unit (Research Triangle Park, NC) at GlaxoSmithKline. 29 All Smyd3 compounds were synthesized by the Cancer Epignetics Performance Unit (Upper Providence, PA) at GlaxoSmithKline.
Compound solids were made up to 5 mM working stocks in DMSO, and a twofold, 12-point dilution series (including vehicle control) was conducted in 384-well plate format (781280, Greiner Bio-One, Monroe, NC). Master plates where stamped using an Apricot liquid handler (Apricot Designs, Covina, CA) into Labcyte ECHO approved low-dead-volume source plates. Using an Echo 555 acoustic dispenser (Labcyte Inc., Sunnyvale, CA), 200 nL of each compound concentration was ejected into 384-well PCR plates (#HSP-3866; Bio-Rad, Hercules, CA) into matching wells. Plates were sealed with adhesive aluminum foil and stored at 4 °C.
Western-Based CETSA
Cells were harvested into 50 mL tubes and centrifuged at 1200g for 5 min. The resulting cell pellets were resuspended to 2e7 cells/mL in serum-free cell culture medium. Fifty microliters of the cells were dispensed into each well of the PCR plate and subjected to a 37 °C to 70 °C heat cycle for 3 min on a Veriti 96-well thermal cycler (Thermo Fisher Scientific, Waltham, MA) with six temperature zones. The heated cells were then equilibrated at room temperature for 3 min before lysis with modified RIPA buffer (#20-188; EMD Millipore) on ice for 10 min. Lysates were centrifuged at 15,000g for 20 min, and clarified supernatants were further processed for Western analysis. Following sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), the separated protein bands were transferred onto nitrocellulose membranes by iBlot (Thermo Fisher Scientific), and the iBind system was used for the anti-Smyd3/anti-IDO1 detection. The integrated band intensities were quantified by Li-Cor densitometry measurement. The thermal curves were analyzed by nonlinear regression using Graph Pad Prism.
EFC CETSA
Cell density was measured with a Vi-Cell-XR Cell Viability Analyzer (Beckman Coulter, Brea, CA) and adjusted to 4e5 cells/mL in DMEM/F12 containing 1% FBS. To each well of the Bio-Rad plates containing prestamped compounds, 12.5 µL cells were dispensed. The resulting final DMSO concentration was 1.6%. The plates were sealed with adhesive foil and incubated with 1200 rpm shaking on a Thermo-Shaker PST-100HL at 37 °C for 2 h. Postincubation, the plates were transferred to a Light Scanner Pro 384-well PCR block (Idaho Technology, Salt Lake City, UT) and heated to 48 °C for 3 min followed by a 1 min cool down to room temperature. A no-heat control plate was set aside and bypassed this thermal cycling event. Fifteen microliters of InCELL Pulse reagent mixture (DiscoverX) was added to the cells at the prescribed ratio (four parts substrate, one part EA reagent, one part lysis detergent). Enzymatic turnover was conducted for 3 h at room temperature in a dark location. The plates were then read using an EnVision 2104 Multilabel plate reader (PerkinElmer, Waltham, MA) operated for ultrasensitive chemiluminescent detection with the following settings: plate type optimized, cross-talk calculated, distance between plate and detector 0 mm, measurement time 0.1 s, glow correction factor 0%.
Cell Health Assays
SMYD3-ePL BacMam transduced HeLa S3 cells were harvested, plated at 1e6 cells per mL, and incubated for 3 h at 37 °C. Plates were sealed using plate seals (AB0580, Thermo Fisher Scientific) and heated to achieve protein melt via the thermal heat cycler (C1000, Bio-Rad). Three temperature gradient (43 °C to 75 °C) melting protocols were compared: (1) 2 min heat gradient followed by 3 min cool down at 25 °C, (2) 3 min gradient followed by 3 min cool down at 25 °C, and (3) pulse cycling with 10 s heat gradient followed by 1 min cool down at 25 °C, repeated 17 times (lid was kept at 40 °C). Plates were unsealed and allowed to cool at room temperature for 20 min. For cell viability evaluation, CellTiter-Glo reagent (G7573; Promega Corporation, Madison, WI) was reconstituted according to manufacturer recommendation, and 5 µL was dispensed to the designated plates. Luminescence (RLU) was quantified via ViewLux (PerkinElmer). For the trypan blue exclusion measure of membrane integrity, 20 µL of cells was pulled from replicate wells in the assay plate and combined with 20 µL of 0.4% trypan blue (15250061; Thermo Fisher Scientific). Twenty microliters of the mix was dispensed into the Cellometer cell counting chamber (Nexcelom Bioscience, Lawrence, MA) for the viability measurement. Percentage viability was recorded for samples melted at specific temperatures.
Data Analysis
Luminescent counts were obtained for each compound at varying concentrations including vehicle control. The data were imported and analyzed in Tibco Spotfire 6.5 (PerkinElmer). Data normalization was conducted as follows:
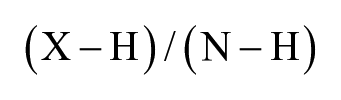
where X is the sample, H is the heated control (DMSO), and N is the no-heat control (DMSO).
Unheated control and heated control values were averaged across plates. Data validation was conducted using mean and median values with little deviation among derived EC50 values. Normalized data values, equivalent to percentage protein remaining in solution, were plotted along the y-axis against the molar concentration for the compounds (x-axis = logarithmic). Using the Spotfire linear regression curve fitting with automatic parameters, EC50 values were obtained for each compound titration.
CETSA for Endogenous SMYD3
Cells were trypsinized, resuspended in serum-containing media to inactivate trypsin, and then washed with phosphate-buffered saline (PBS) and resuspended in serum-free media to 40e6 cells/mL. Fifteen microliters of this suspension (600,000 cells) was added to each well of a 96-well PCR plate containing 5 µL of serially diluted compounds or DMSO in serum-free media (final DMSO concentration 0.1%). Cell suspensions with DMSO for no-heat denaturation controls were prepared similarly in parallel in PCR strip tubes. Compound or DMSO cell suspension mixtures were incubated at 37 °C, 5% CO2, for 1 h. Afterward, the PCR plate was placed in a thermocycler preheated to the appropriate temperature (48 °C for MDA-MB-453 and 49 °C for SK-OV-3, determined experimentally previously for each cell line, not shown) for heat denaturation for 3 min, followed by 3 min rest at room temperature. The heated PCR plate and unheated strip tubes were then placed on ice, and cells were lysed by the addition of 20 µL 2X modified RIPA buffer (#20-188, EMD Millipore) containing 2X Thermo Halt Protease Inhibitor Cocktail (#78430, Thermo Fisher Scientific) and 2 µL/mL Benzonase (#E8263, Sigma-Aldrich). Samples were vortexed vigorously and then incubated on ice for 20 min to allow complete cell lysis. Lysates were then transferred to prechilled, low-retention microfuge tubes and centrifuged at 21,000g for 20 min at 4 °C. Supernatants were transferred to new microfuge tubes and subjected to SDS-PAGE and Western blotting with 1:5000 rabbit anti-SMYD3 (ab183498, Abcam) and 1:50,000 mouse anti-Tubulin (#T9026, Sigma-Aldrich) antibodies. SMYD3 band intensities were quantified with Odyssey software (LI-COR, Lincoln, NE) normalized to tubulin band intensities and plotted relative to 100% unheated DMSO control.
Smyd3 In-Cell Western Assays
For MEKK2-K260 trimethyl In-Cell Western, HEK293 cells (ATCC, CRL-1573) were cultured in RPMI (A10491; Life Technologies, Waltham, MA) supplemented with 10% FBS (Sigma-Aldrich, 12176C). Cells were transfected in flasks by combining HA-tagged MEKK2 plasmid DNA (pLEX/HA-MEKK2, GRITS:132173) and SMYD3 plasmid DNA (pLenti-SMYD3) or empty plasmid (pcDNA3.1+, Invitrogen #V90-20) in Opti-MEM reduced serum media (Thermo Fisher Scientific, 51985091) followed by the addition of lipofectamine 2000 (Thermo Fisher Scientific, 11668019). Flasks were then incubated for 4 h prior to seeding cells in 384-well clear-bottom plates (Greiner Bio-One, 781956) and immediately dosed with compound to achieve a 20-point, twofold dilution scheme, with a final DMSO concentration of 0.15%. After a 24 h incubation, all media were aspirated and cells were fixed and permeabilized by adding 50 µL of ice-cold 100% methanol followed by PBS washing and blocking in Odyssey blocking buffer (Li-Cor, 927-40000). Primary antibodies including anti-HA (Covance, MSS-10 IR HA.11, 16B12) and rabbit anti-MEKK2-K260me (custom YenZym, YZ5047 first cycle, 0.14 mg/mL) were prepared together in Odyssey blocking buffer + 0.1% Tween-20 and incubated on cells for 16 h at 4 °C followed by washing. Secondary antibodies in Odyssey blocking buffer + 0.1% Tween-20 (goat anti-mouse 680LT; Li-Cor, 926-68020, and goat anti-rabbit 800CW, LI-COR, 926-32211) were incubated on cells for 1 h followed by washing and imaging. Plates were imaged using the Odyssey Imager (Li-Cor) and signal intensity calculated. Any compound-treated well with less than 70% of the positive control HA signal was omitted from analysis because of cell loss. The ratio of MEKK2-K260me to MEKK2-HA was plotted versus compound concentration and fit to a standard four-parameter logistic model to calculate EC50. All data represent a minimum of two separate biological replicas with an EC50 range of less than threefold.
Results and Discussion
A key requirement for the broad deployment of HTDR-CETSA across target classes was to establish a robust method for expression of the ePL fusion target protein in a cellular context. Our primary strategy was to genetically incorporate the ePL tag at the C-terminus of the protein of interest and transduce into a relevant screening assay cell line using the BacMam system. 30 The initial protein target chosen for the development of the HTDR-CETSA assay was SMYD3, a human histone-lysine N-methyltransferase. Full-length, human SMYD3 with a C-terminal ePL fusion tag was overexpressed in HeLa S3 cells using BacMam transduction. BacMam titration enabled tunable control of the SMYD3-ePL fusion protein levels to nearly match the endogenous SMYD3 expression ( Fig. 1A ). Next, we compared the thermal stability of the endogenous SMYD3 (determined by Western readout) to the melt profile of the overexpressed SMYD3-ePL (InCELL Pulse detection), to confirm that the ePL tag does not appear to perturb the thermal stability of endogenous SMYD3 ( Fig. 1B ). The Western-based thermal shift assay included a centrifugation step to remove the insoluble protein aggregates, whereas the EFC thermal shift assay did not. This strongly implies that the ePL tag is rendered inaccessible because of thermally induced, irreversible protein denaturation. It also provides strong evidence that the 42 amino acid long ePL does not have a fold of itself that could contribute to the thermal transition of the SMYD3-ePL compared with untagged SMYD3. Furthermore, we show that compound GSK-1, a tool compound previously shown to positively stabilize the thermally induced unfolding of recombinant SMYD3 by ThermoFluor ( Suppl. Fig. S1 ), stabilized both endogenous SMYD3 and the SMYD3-ePL fusion protein to a similar extent. In addition, knowing that the InCELL Pulse detection methodology uses a detergent-based lysis, this experiment demonstrated that the detergent does not keep “melted” SMYD3-ePL solubilized in the supernatant, which would result in the undesirable production of a spurious background signal.

Development of enzyme fragment complementation readout for the cellular thermal shift assay () and in the presence of a tool compound (■ ePL,  Western). () and in the presence of a tool compound (■ ePL, Western).
Western). () and in the presence of a tool compound (■ ePL, Western).
A second, unrelated target, IDO1, was chosen to evaluate the broader applicability of this assay. Indoleamine 2,3-dioxygenase (IDO1), a monomeric heme-containing enzyme, catalyzes the initial rate-limiting step in the catabolism of tryptophan via the kynurenine pathway and is stimulated in numerous cell types in response to proinflammatory cytokines. 31 BacMam titration of IDO1-ePL fusion construct also demonstrated tunable control of expression levels and furthermore recapitulates secondary biology, as endogenous IDO1 expression is dependent on IFNγ ( Fig. 1C ).
The utility of the BacMam ePL approach was again demonstrated, as evidenced by the near equivalency of the IDO1-ePL EFC-based detection compared with a Western readout for determination of the ΔTm ( Fig 1D ), as well as tool compound (GSK5628) induced thermal stabilization of the protein target ( Table 1 ). 29 In summary, data generated for the two model systems presented, as well as unreported data for five additional internal discovery efforts spanning multiple protein target classes, indicate that fusion of the ePL tag to the target protein C-terminus does not typically alter the Tm of the tagged target protein relative to the endogenous cellular counterpart nor interfere with the magnitude of thermal stabilization induced by drug molecule interaction. Importantly, upon heat-denatured aggregation, the ePL subunit is not competent to participate in enzyme fragment complementation; thus, no spurious chemiluminescent signal is observed in the absence of soluble, properly folded target-ePL protein. Finally, assay signal was unaffected by the presence of low-serum media, residual test compound, or uncentrifuged cellular debris components. These data confirmed that the extensive washing, centrifugation, and detergent-free liquid N2 freeze-thaw lysis steps of the original CETSA protocol could be eliminated. Collectively, these findings indicated that this homogenous reporter system could be integrated and adapted for streamlined quantification of soluble protein in the HTDR-CETSA platform.
Summary of Tm Determination in the Absence or Presence of the IDO1 Tool Compound, GSK5628, Determined by Either Western or ePL Detection.
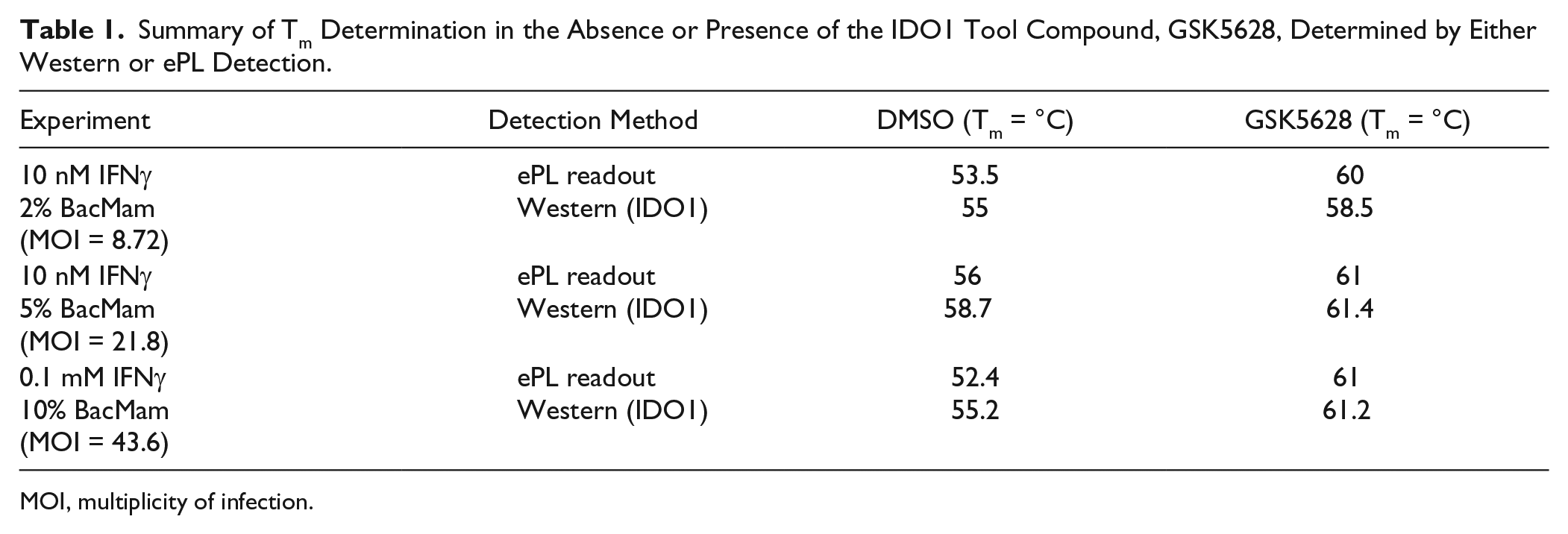
MOI, multiplicity of infection.
Having established a system for expression of the ePL reporter across target classes and demonstrating that this chemiluminescent InCELL Pulse assay is equivalent to the endogenous Western-based CETSA readout, we miniaturized the assay into a 384-well plate-based format. We experimentally determined that a 0.2% BacMam transduction (MOI = 0.35) of Smyd3-ePL using 5000 to 10,000 cells ( Fig. 2A ) gave a robust signal-to-noise ratio (S/N) for ePL detection. We empirically optimized assay volume by incubating various quantities of HeLa S3 cells expressing Smyd3-ePL with DiscoverX InCELL Pulse reagent mixture held at fixed ratios and monitoring chemiluminescent signal intensity. Assay signal intensity increased linearly with increasing assay volume ( Suppl. Fig. S2 ). Acceptable performance was obtained using 3 to 12.5 µL cells per assay well (1200–5000 cells in a final assay volume of 6.6–27.5 µL), suitable for 384-well or 1536-well PCR plate format capabilities. The EFC signal continued to increase in intensity past the recommended 30 min read time and did not reach a signal plateau until 2 to 3 h after reagent addition. Improved homogenization via continuous mixing using a plate shaker, or repeated aspiration/dispense steps on a liquid handler, did not affect the latency of signal production ( Suppl. Fig. S2 ). Critically, ePL signals for Smyd3 samples heated above their Tm did not increase beyond baseline during this time window, indicating that this signal latency is not attributable to restoration of functional competence of the aggregated ePL tag ( Fig. 1B ). Rather, these findings are consistent with slow association of the EA subunit with the ePL fusion tag. 32 To maximize S/N and optimize the assay window, the EFC detection reaction for HTDR-CETSA profiling was allowed to reach completion for 3 h at room temperature in a dark location.
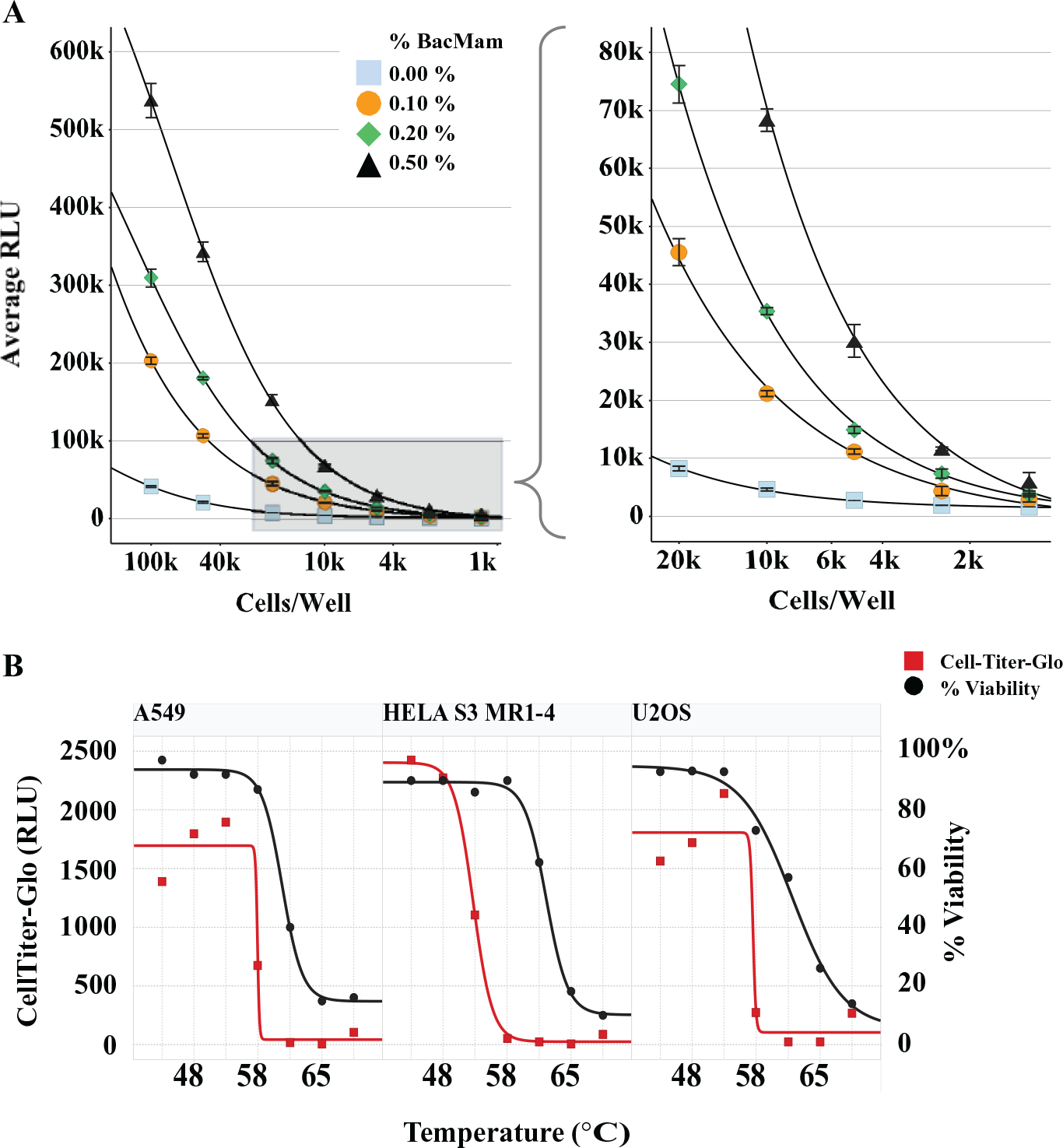
High-throughput dose-response cellular thermal shift assay (CETSA) parameter optimization. (
Whole cell-based CETSA assays are dependent on maintenance of cellular membrane integrity during the compound treatment and heating regimen, ensuring that cellular permeability is not altered, and the observed pEC50 values for target engagement are reflective of true intracellular compound concentrations at equilibrium. We characterized cell membrane integrity of three assay cell lines in response to the CETSA heat cycle using trypan blue dye exclusion. We calculated an approximate trypan blue XC50 of 63 °C for A549 cells and 64 °C for the HeLa and U2OS cells ( Fig. 2B ). These values are consistent with previous reports and indicate that membrane integrity of these cell lines is preserved at temperatures above the Tm of typical soluble intracellular proteins.3,4
In addition to cell membrane integrity, it is of importance to consider cell health and viability during CETSA heat cycles. Although the short time frame for heating and cell lysis steps is likely insufficient to elicit the full heat-shock response, even abbreviated heat cycles of 0.5 to 1 min can have immediate detrimental effects on cells, including increased necrosis and diminished viability. 33 As a secondary means of characterizing cell viability in response to the CETSA heat cycle, the three cell lines were also analyzed using CellTiter-Glo (CTG), which measures adenosine triphosphate (ATP) production as a function of the metabolic health of the cell. The CTG curves were used to calculate an approximate metabolic cell death XC50 of 53 °C for HeLa S3 cells and 58 °C for both the A549 and U2OS cell lines ( Fig. 2B ). Examination of the most commonly applied CETSA heat regimens comparing 2 min and 3 min fixed cycles to a pulsed cycle in HeLa S3 cells revealed only minor differences in CTG (53–55 °C) and trypan blue (63–65 °C) cell death XC50 values ( Suppl. Fig. S3 ). This illustrates the variability in cell health and membrane integrity in response to an increase in temperature and highlights the need to understand the relationship between cell death, membrane integrity, and target protein melting temperature in a given cell line of interest as part of selection criteria for the isothermal heat regimen.
To probe the utility of our HTDR-CETSA assay in linking biochemical potency to cellular efficacy by directly measuring cellular target engagement, we incorporated this assay for compound profiling in the lead optimization stage for a recent SMYD3 discovery program at GSK. SMYD3 is a lysine methyltransferase involved in the regulation of gene transcription through methylation of histone and nonhistone proteins. SMYD3 is overexpressed in several cancers including breast, liver, and rectal carcinomas.34,35 Knockdown of SMYD3 expression with siRNA resulted in significantly suppressed growth in several cancer cell lines, suggesting that SMYD3 is essential for tumor proliferation. 34 These studies highlight the involvement of SMYD3 in cancer and suggest an opportunity for chemotherapeutic intervention, yet the molecular mechanism of SMYD3 in cancer remains complex and not well understood. SMYD3 has been reported to methylate multiple histone lysines including lysine 4 in histone H3 (H3K4), lysine 5 in histone H4 (H4K5), and lysine 20 in histone H4 (H4K20).34–37 More recently, SMYD3 has been shown to regulate the MEK/ERK signaling pathway by methylation of mitogen-activated protein kinase kinase kinase 2 (MEKK2) at K260 and loss of SMYD3 catalytic activity inhibited tumorigenesis in the presence of oncogenic Ras. 38 This regulatory complexity highlights the inherent difficulty in the development and implementation of cellular mechanistic assay(s) that are predictive of a SMYD3-dependent cancer phenotype on the time scale of a typical discovery program at GSK.
We generated isothermal (48 °C) dose-response curves for 123 compounds originally identified as potent SMYD3 inhibitors (pIC50 > 6.9) in a biochemical scintillation proximity assay screen that measured SMYD3 methyltransferase activity, in the presence of cofactor (S-adenosyl methionine) and substrate (recombinant MEKK2). Compounds were profiled in duplicate as 12-point full-curve dilution series in Smyd3-ePL transduced HeLa S3 suspension cells, and HTDR-CETSA pEC50 values were generated using linear regression analysis. Coefficient of variance (CV) measurements indicate excellent assay reproducibility: <15% CV was observed between duplicate pEC50 determinations for ~90% of the compounds tested (109/123). Of the CV outliers, 12 of 14 cluster at pEC50 values <4.0, which we have defined as the lower threshold indicative of cell-active Smyd3 inhibitory compounds ( Fig. 3A ). To further establish assay robustness, we also examined the effect of heating temperature on ITDR-CETSA EC50 values. To accomplish this, eight compounds were tested using HTDR-CETSA at four temperatures spanning from 46 °C to 52 °C in quadruplicate. Compound rank ordering of pEC50 CETSA values is largely preserved regardless of the temperature chosen for the ITDR-CETSA experiment ( Fig. 3B ). As might be expected, increasing temperatures lead to lower CETSA pEC50 values likely because of the irreversible mechanism of protein unfolding and faster compound off-rates associated with elevated temperatures. These observations are consistent with a proposed equilibrium two-state model, which assumes a single-site interaction exclusively with the native protein and a two-state protein unfolding transition. 39 These data establish that minor deviations in heating temperature are unlikely to negatively influence reproducible rank ordering of potency.
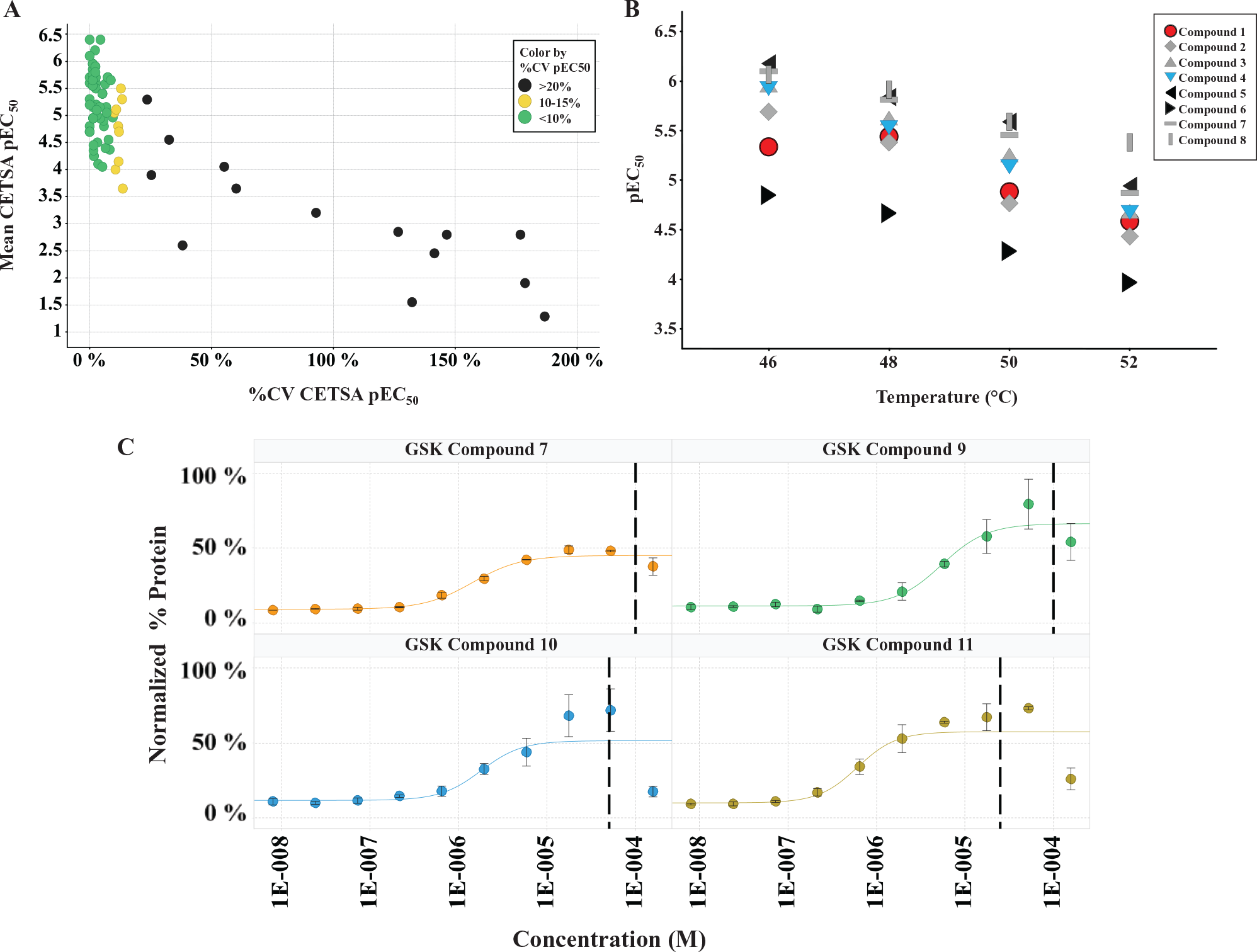
Smyd3-ePL compound profiling by high-throughput dose-response cellular thermal shift assay (HTDR-CETSA). One hundred twenty-three compounds identified as potent inhibitors in a biochemical screen were profiled in duplicate as 12-point full-curve dilution series in HeLa S3 suspension cells. Fitted mean pEC50 values and coefficients of variance (CVs) are plotted and indicate <15% CV for 90% of the compounds tested (97% of the cell-active compounds pEC50 >4.0); the CV outliers cluster at pEC50 values <4.0 (cell inactive). (
Atypical curve behavior at the highest dose concentrations was observed for many of the compounds profiled; this was evidenced as a protein destabilization phenomenon ( Fig. 3C ). These deviations complicated automated curve fitting and necessitated manual inspection and removal of outlier data points. Poorly soluble compounds are a common nuisance in drug discovery. These may take the form of reversible colloidal aggregates, which spontaneously form in aqueous biochemical buffers and cell culture media at micromolar concentration. In cell culture, these colloids appear to act as reservoirs that reduce the free, effective concentration of the drugs, diminishing their true efficacy. 40 In the case of CETSA, this would be predicted to result in reduced target engagement and diminished thermal stabilization. To further investigate these observations, we empirically determined aggregation concentration for select compounds using the Corning EPIC, a high-throughput plasmon waveguide resonance instrument platform that can directly measure solubility-driven solution aggregation phenomena. In all instances, the CETSA destabilization effect we observed correlated with doses exceeding the aggregation concentration measured by Corning EPIC. This important caveat must be taken into account when choosing the maximum concentration range for determining EC50 values for HTDR-CETSA. Compound aggregation considerations are especially critical when using CETSA screening to run large campaigns at a singular saturating concentration, deemed “single shot” in the industry, as these potential candidates would systematically read as false-negatives and would be excluded from further progression.
SMYD3 has been demonstrated to methylate MEKK2 at lysine K260. Cellular mechanistic assays were developed to assess the potency of SMYD3 inhibitors on this substrate in vitro. Correlations between HTDR-CETSA pEC50 values with the cellular MEKK2 assay pEC50 values (R2 = 0.49; 82 compounds) indicate that quantitative cellular target engagement data can be predictive of a SMYD3-dependent cellular mechanism ( Fig. 4A ). In addition, a subset of 15 compounds with an alternative mode of inhibition also showed a good correlation (R2 = 0.82) between HTDR-CETSA and the MEKK2 cellular mechanistic assay ( Fig. 4B ). Compared with the MEKK2 assay, CETSA pEC50 values were systematically right shifted approximately 0.5 log units across the pEC50 range of 5.5 to 7.0, although rank ordering was largely preserved. These data highlight the ability of HTDR-CETSA to quantitatively assess target engagement and its comparability to the cellular mechanistic readout. HTDR-CETSA was accomplished using a less engineered, highly simplified assay format with greater throughput and same-day turnaround.
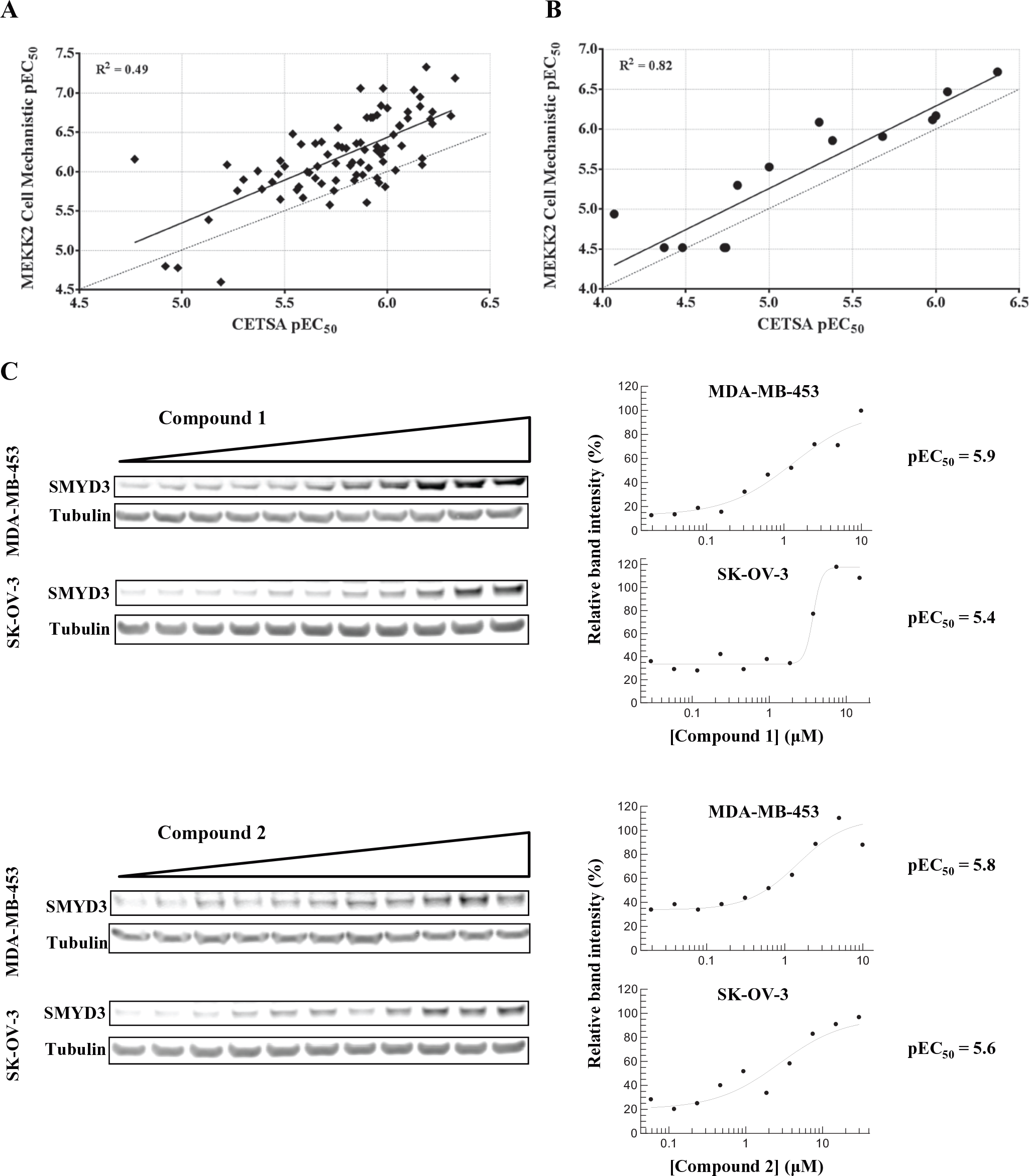
Correlation of high-throughput dose-response cellular thermal shift assay with an in-cell Western MEKK2 K260 cellular mechanistic assay applied to (
Quantitative measures of target engagement in HeLa S3 or other workhorse assay cell lines by HTDR-CETSA are of value only if they correspond to directly translatable measures of target engagement and inhibition of the endogenous protein target in disease-relevant cells. To demonstrate this, we profiled select Smyd3 inhibitors identified by HTDR-CETSA in both MDA-MB-453 (breast metastatic carcinoma) and SK-OV-3 (ovary adenocarcinoma) cancer-relevant cell lines ( Fig. 4C ). Data shown for two inhibitors profiled by traditional Western-based ITDR-CETSA using an anti-Smyd3 antibody confirmed dose-dependent endogenous Smyd3 target engagement, producing pEC50 values of ~5.9 (MDA-MB-453) and ~5.5 (SK-OV-3). These are in close agreement with HeLa S3 HTDR-CETSA–derived values of pEC50 = 6.1 for both compounds. Additional compound profiling further confirmed concordance between the two assays (data not shown).
In summary, we have described a miniaturized, single-pot, HTDR-CETSA capable of quantitative, direct cellular target engagement measurements in a minimally engineered system with no antibody requirements ( Fig. 5 ). By combining the tunability of the BacMam system with a robust chemiluminescent reporter (DiscoverX EFC technology), we initially demonstrated the utility of this assay in a 384-well format for compound profiling on SMYD3. 41 We show that a single, quantitative measure of cellular target engagement correlates well with cellular mechanisms, providing an opportunity for improving clinical translation through integration of HTDR-CETSA into the lead optimization phase for early drug discovery programs.
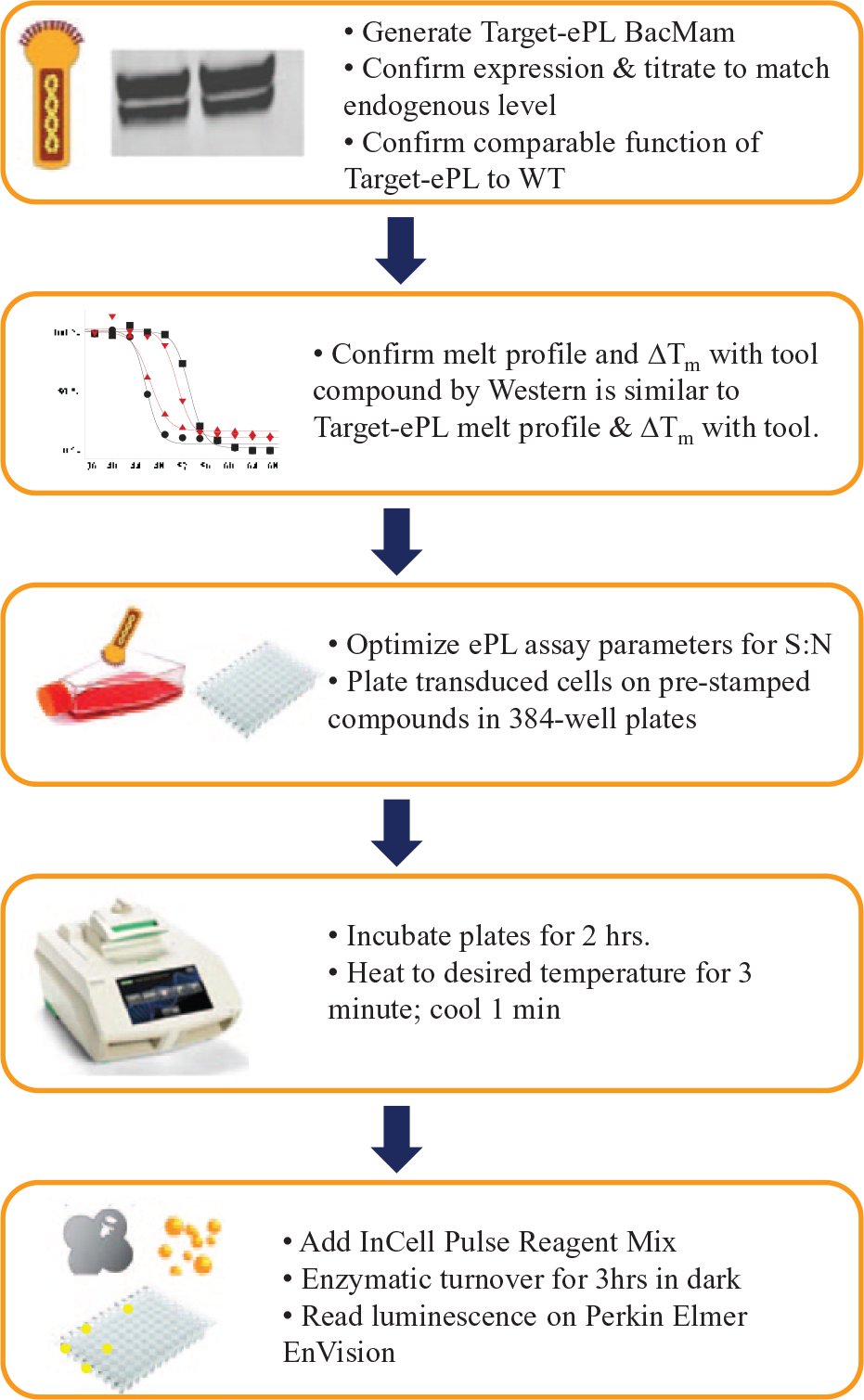
Scheme for high-throughput dose-response cellular thermal shift assay (HTDR-CETSA) target progression. Target-ePL BacMam is first generated, expression confirmed, and then closely matched to endogenous target expression levels by BacMam titration. Functional activity of target ePL should be tested for comparable activity to wild type. Experiments are conducted to confirm that target-ePL melt profile and ΔTm with a tool compound as determined by ePL readout is nearly identical to wild-type melt profile and ΔTm with a tool compound as determined by Western. Assay conditions are optimized (i.e., cell type, cell density, and detection reagents). The assay is then progressed for HTDR-CETSA compound profiling, with BacMam target-ePL transduced cells dispensed onto 384-well Bio-Rad PCR plates containing prestamped compounds.
Implementation of the EFC detection platform allows introduction of a generic, target-independent readout for protein quantification; we anticipate that other assay platforms based on engineering proteins with generic signaling tags such as time-resolved fluorescence resonance energy transfer, 42 bioluminescence resonance energy transfer, 43 or NanoLuc 44 may be readily adaptable to HTDR-CETSA–type screening. In early drug discovery campaigns against novel targets, these modalities may prove ultimately more practical to implement than antibody-based protein quantification strategies such as Alpha-Screen, which requires pairs of well-characterized, highly specific, noncompeting antibody reagents for each new protein analyte. Adapting CETSA for high throughput using antibody-based homogenous detection has proven problematic even in ideal cases, as significant and unexpected ligand-induced signal suppression has been observed due to loss of antibody recognition in the presence of ligand.4,45 Although our focus is primarily the determination of target engagement in a cellular setting, the assay platform we describe can be readily adapted for lysate measurements, which would serve as a counterscreen for compounds found to be cell inactive because of a lack of cellular permeability or restricted subcellular access to the target.
Measures of direct target engagement by HTDR-CETSA can complement or potentially replace multiple cellular mechanistic assays and provide a fast track to test only the most promising candidates in expensive and often difficult-to-obtain human disease-relevant primary cells and animal models. It does, however, remain to be seen how widely applicable these findings are across diverse target classes; to that end, at GlaxoSmithKline, we have recently applied HTDR-CETSA with success to five early discovery projects. In addition, DiscoverX has recently presented on the commercialization of their InCELL Pulse Platform for use across the broader scientific community. 46
Future applications for HTDR-CETSA include screening of membrane targets 24 and further miniaturization into 1536-well plates for full chemical diversity screening in hit identification. 45 Accessing recent advances in genome engineering (CRISPR), one can envision the stable integration of target-ePL fusions in the proper genetic loci into whole organisms; this could give us the ability to directly screen in isolated primary cell types or perhaps even to directly measure preclinical target engagement in tissues at the relevant site of action. We predict that the ability to measure and gain a detailed understanding of engagement of a drug with its molecular target at the earliest stages of drug discovery will reduce attrition and improve success in the clinic.
Footnotes
Acknowledgements
For his intellectual insights and patience in exploring the utility of DiscoverX’s enzyme fragmentation complemention technology, the authors would like to thank Daniel Treiber.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.