
Abstract
The level of mechanistic understanding required for drug discovery is a central feature of most strategies. However, an understanding of mechanism is not required for regulatory approval. This paradox is particularly relevant to the role of phenotypic assays in drug discovery. A recent analysis revealed that phenotypic drug discovery strategies were more successful for first-in-class medicines, whereas target-based molecular strategies were more successful for followers (Nat. Rev. Drug Discov.
Keywords
Introduction
It is helpful to understand how something works in order for it to be repaired or improved. Understanding how a clock, computer, or automobile works is important to fix them when broken or improve their operation. In contrast, little understanding of the details of how they work is required for them to be used effectively. This is analogous to medical research, with the caveat that drug mechanisms are known at different levels of resolution. Drug mechanism can be understood at the whole organism level, the level of a cell, the level of a biochemical pathway, and the molecular level. Medical research and drug discovery use the understanding of mechanism to help discover new treatments, while regulators and patients are concerned with if the medicine works, if the medicine is safe, and how to use the medicine.
This paradox is further complicated by the differing definitions and use of mechanism in drug discovery. The initial identification of a chemical to be developed into a medicine generally occurs by screening molecules in a biological assay relevant to human disease. These biological assays are established using available mechanistic knowledge to reflect some aspect of the disease. Ideally, the outcomes from the assay will translate to clinical disease and identify molecules that are both efficacious and safe. Current drug discovery strategies include molecular approaches and empirical approaches. The molecular approaches tend to be predominantly hypothesis driven and are generally referred to as target based due to the focus on modulation of the activity of a specific gene product. In practice, the gene product or target is considered the key feature of a drug’s mechanism of action. The empirical approach is referred to as phenotypic due to its reliance on phenotypic measures of response in assays that are developed to be physiologically relevant, such as animal and cellular models. In these assays, there are less assumptions regarding the mechanism of action.
A recent analysis of drug discovery success has revealed that empirical phenotypic screening has had more success in providing starting points for first-in-class small-molecule new molecular entities (NMEs) than target-based approaches. 1 Discovery strategies and the molecular mechanism of action (MMOA) for NMEs and new biologics that were approved by the U.S. Food and Drug Administration (FDA) between 1999 and 2008 were analyzed. 1 Of the 259 agents that were approved, 75 were first-in-class drugs with new MMOAs, and out of these, 50 (67%) were small molecules and 25 (33%) were biologics ( Fig. 1 ). The results also showed that the contribution of phenotypic screening to the discovery of first-in-class small-molecule drugs exceeded that of target-based approaches—with 28 and 17 of these drugs coming from the two approaches, respectively—in an era in which the major focus was on target-based approaches.
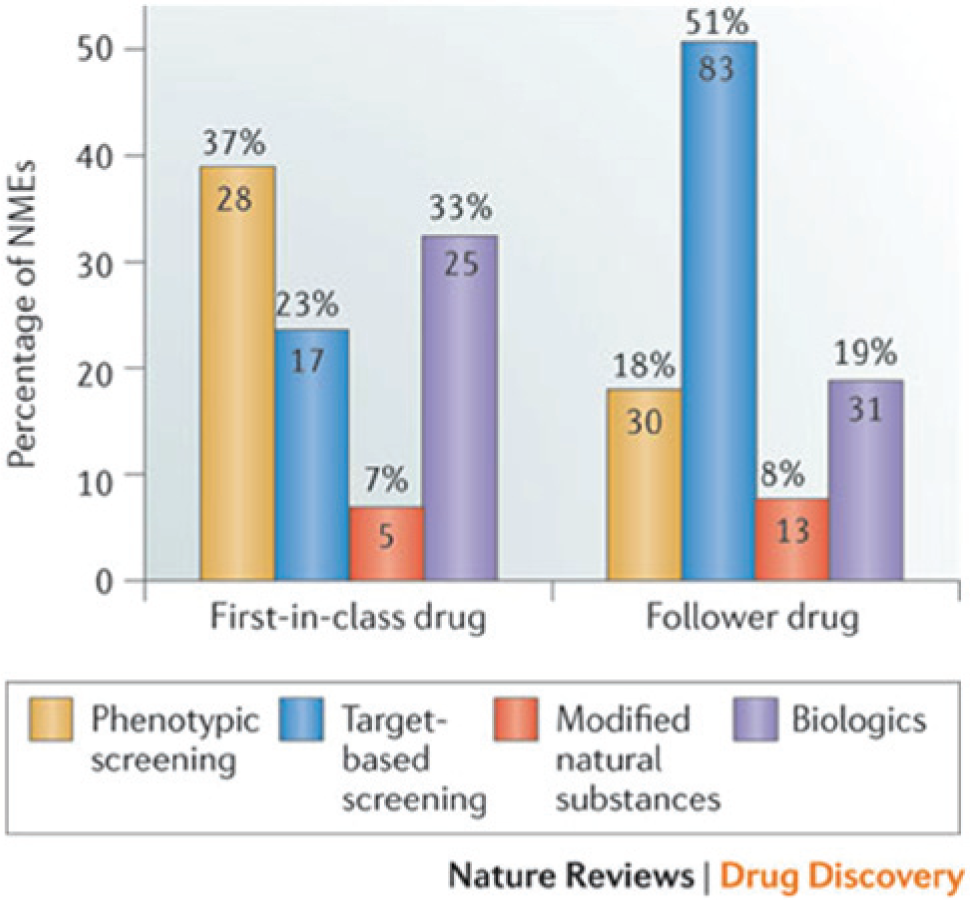
The distribution of new drugs discovered between 1999 and 2008, according to the discovery strategy. The graph illustrates the number of new molecular entities (NMEs) in each category. Phenotypic screening was the most successful approach for first-in-class drugs, whereas target-based screening was the most successful for follower drugs during the period of this analysis. The total number of medicines discovered via phenotypic assays was similar for first-in-class and follower drugs—28 and 30, respectively—whereas the total number of medicines discovered via target-based screening was nearly five times higher for follower drugs versus first-in-class drugs (83 to 17, respectively). From Swinney and Anthony. 1 Reprinted with permission.
Examples of first-in-class medicines discovered by phenotypic screening included those discovered using animal models, such as ezetimibe (Zetia) for reducing levels of blood cholesterol and those discovered with cellular assays, such as vorinostat (Zolinza), the first HDAC inhibitor, that was reported to come from the observation that DMSO had an unexpected effect on cancer cells, and linezolid (Zyvox), an oxazolidinone antibiotic identified in bacterial assays. Target-based successes included a number of tyrosine kinase inhibitors for cancer, including gefitinib (Iressa) (epidermal growth factor receptor), imatinib (Gleevec) (BCR-ABL), sorafenib (Nexavar) (Raf), and sunitinib (Sutent) (vascular endothelial growth factor receptor/platelet-derived growth factor receptor); antivirals include maraviroc (Celsentri) (CCR5), raltegravir (Isentress) (HIV integrase), and zanamivir (Relenza) (influenza neuraminidase).
This analysis proposed that lower productivity partly reflected target-based discovery’s lack of consideration of the molecular complexities of how the drugs work. Knowing the parts of an efficient machine—a watch, an automobile, or a computer—is not enough to describe how it works. The parts must collaborate in precise ways to provide accurate time, reliable transportation, or processed information.
Biology is exponentially more complex. The phrase “molecular mechanism of action” describes the way that biological parts collaborate to provide an effective and safe medicine. Addressing the MMOA would contribute to reversing the low productivity of target-based discoveries, because merely knowing the identity of a part involved in a defect may not be sufficient to repair a malfunctioning machine. We postulated that a target-centric approach for first-in-class drugs, without consideration of an optimal MMOA, may contribute to the current high attrition rates and low productivity in pharmaceutical research and development.
These observations led to the proposal that the progression of drug discovery from unmet medical need to best-in-class medicines is facilitated by the use of phenotypic assays to identify first-in-class medicines and their respective MMOAs. Progression correlates with an iterative increase in knowledge to specifically address a phenotype related to the unmet medical need. Early in the progression, the knowledge is achieved by empirical analysis. Ideally, as more knowledge is gained to link a specific mechanism of modulation (MMOA) to the desired phenotype, drug discovery can focus efforts toward addressing specific hypotheses ( Fig. 2 ). 2
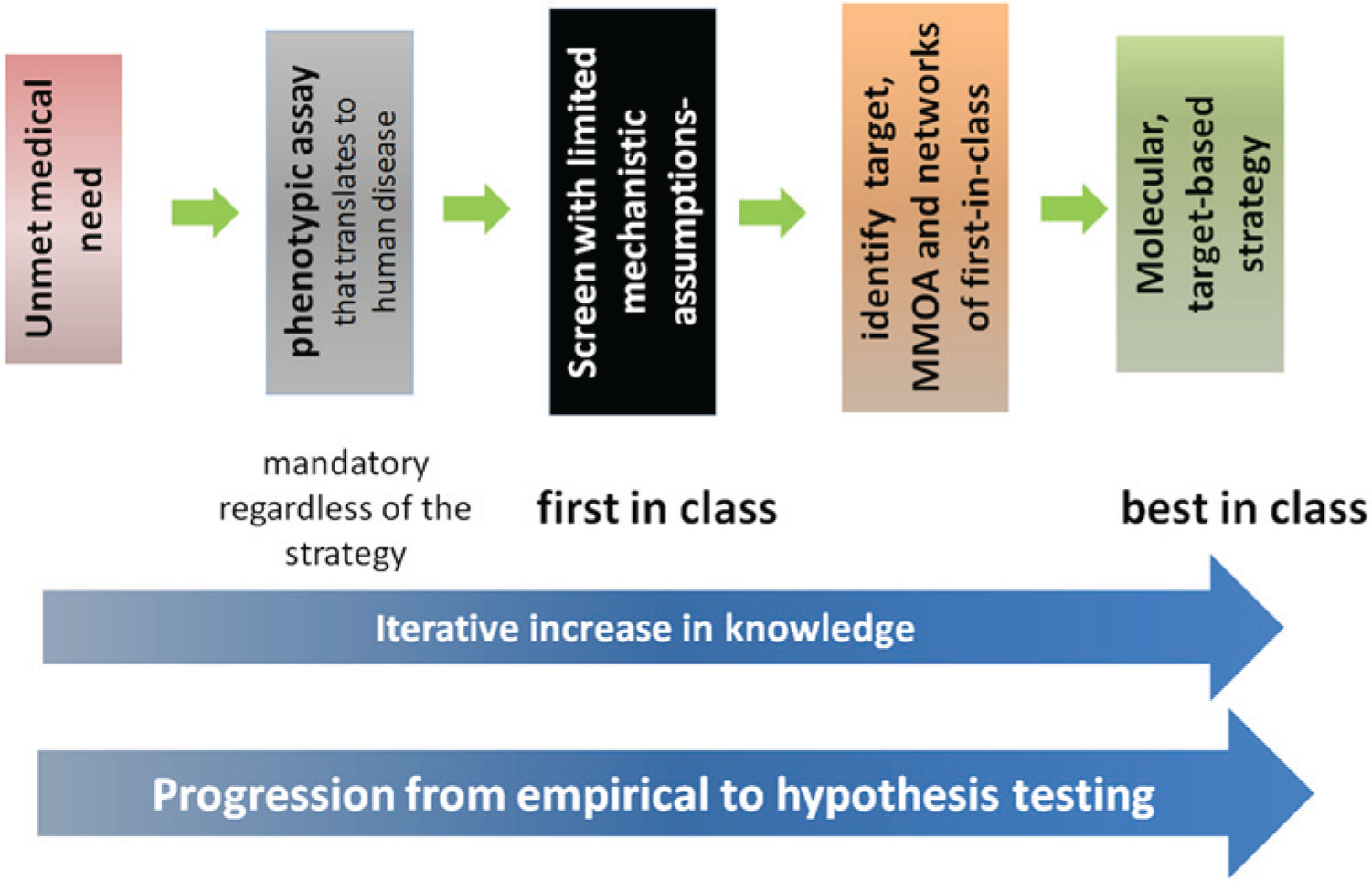
Progression of drug discovery from unmet medical need to best-in-class medicines. This figure is an oversimplified schematic highlighting the contribution of empirical approaches to first-in-class medicines, hypothesis-driven approaches for best-in-class medicines, and the role of mechanistic understanding. Progression correlates with an iterative increase in knowledge to specifically address a phenotype related to the unmet medical need. Early in the progression, the knowledge is achieved by empirical analysis. Ideally, as more knowledge is gained to link a specific mechanism of modulation to the desired phenotype, drug discovery can focus efforts to address specific hypotheses. MMOA, molecular mechanism of action. From Swinney. 2 Reprinted with permission.
However, there is a reluctance to reintroduce the phenotypic strategy into drug discovery and to move compounds forward into development that were discovered using assays where the target is unknown or mechanism of action is not clearly defined. This reluctance highlights the mechanistic paradox. Not knowing the target or MMOA casts doubt on the translatability of the phenotypic assays to human disease. Tools to increase the translatability, such as patient-specific stem cells, capture some of the mechanistic aspects. But it is a major challenge for the mechanistic aspects captured by the stem cells to create opportunities for medicinal chemists. How is this gap bridged?
Addressing this paradox requires the understanding that mechanism has different meanings and implications depending on context and intent. For example, different types of mechanistic information are helpful to a medicinal chemist working to design molecules versus a pharmacologist developing translational assays. At the other extreme, a patient rarely considers “how a drug works” and is concerned primarily with whether the drug will be effective and how to use the drug. Different communities of expertise in biomedical research will have different requirements for mechanistic understanding and, consequently, different definitions.
The intent of this article is to address the relationship between the mechanistic paradox and phenotypic screening. The specific questions asked were (1) what type of phenotypic assays were used as starting points, and (2) what type of mechanistic information was used in the format of the assays? To address these questions, the first-in-class medicines discovered with phenotypic assays approved between 1999 and 2008 and those approved in 2012 were analyzed with respect to the assay formats and the mechanistic knowledge available to choose the starting points. To facilitate the analysis, the NMEs were categorized separately with respect to assay formats and the mechanistic understanding that informed the assay formats.
Assays Formats Used for Successful Phenotypic Drug Discovery
The lead molecules were discovered using animal models, cellular assays with a functional marker, cellular assays with a pathway/biochemical marker, and clinical serendipity ( Table 1 ).3–36 The categorization of molecules between cells with a functional marker and cells with a pathway was chosen to differentiate between the use of a functional marker and a biochemical marker—for example, cell death and/or differentiation and viral infectivity were considered functional markers, whereas measurement of calcium flux, membrane potential, and accumulation of substrate were considered biochemical markers of activity. Most first-in-class small molecules discovered between 1999 and 2008 using a phenotypic strategy were found using animal models and cellular assays with a functional response. The molecules identified in animal models were associated with metabolic diseases (lipids and diabetes), CNS (anticonvulsants), pain, and inflammation. Cellular assays with a functional marker such as cytotoxicity were successful for infectious diseases and cancer. In this period, three medicines were discovered using cellular assays with a pathway/biochemical analysis (caspofungin/glucan biosynthesis, cinacalcet/increase in calcium signaling in bovine parathyroid cells, and miglustat/glycolipid biosynthesis), and two medicines were discovered with clinical serendipity (memantine’s CNS effects were observed during clinical trials designed to evaluate its antiviral potential and sinecatechins are a component of green tea).
Phenotypic Assay Strategies That Led to the Initial Discovery of Candidates Where a First-in-Class New Molecular Entity Was Ultimately Approved by the U.S. Food and Drug Administration.
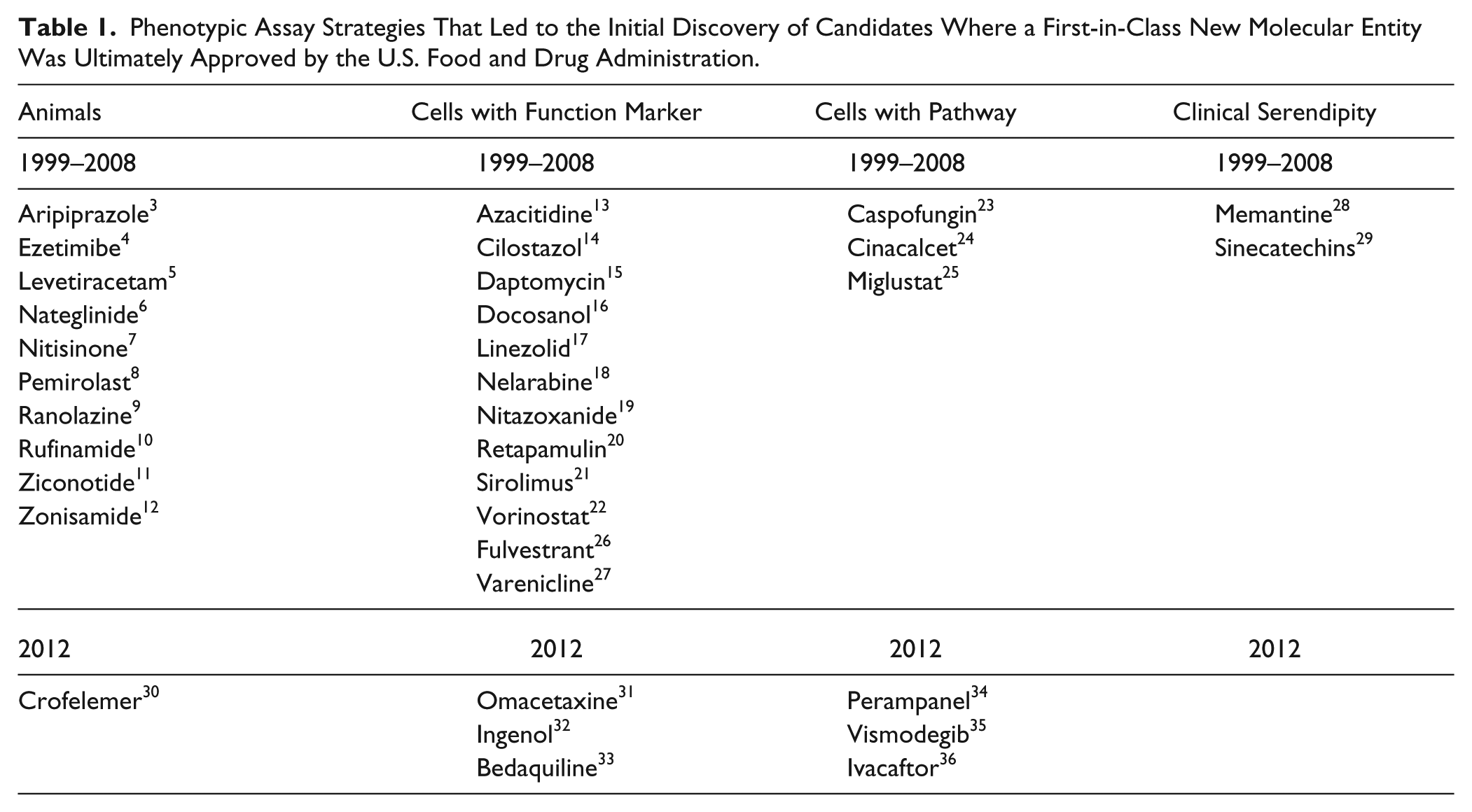
Analyzing NMEs approved by the U.S. FDA in 2012, 7 of the 39 were identified as first-in-class small molecules discovered with phenotypic assays ( Table 1 ). Comparison to 1999–2008 showed a relative increase in those discovered using cell assays with a pathway/biochemical approach. Three (perampanel, vismodegib, and ivacaftor) of the 7 were discovered with a pathway approach compared with only 3 of 28 over the 10-year span between 1999 and 2008. Perampanel was discovered in assays that measured AMPA-induced cortical neuron cell death and calcium influx, 34 vismodegib was discovered as an activator of the hedgehog pathway, 35 and ivacaftor was discovered in cell-based assay of membrane potential looking for either potentiators or correctors in NIH/3T3 cells expressing F508-CFTR. 36 This may represent a more successful integration of empirical and molecular strategies.
It is also of interest to note details of the discoveries of bedaquiline and omacetaxine with respect to phenotypic discovery. Omacetaxine was originally identified more than 35 years ago as a novel plant alkaloid with antitumor properties. 31 Its mechanism of action was found to be inhibition of protein translation. Its clinical development was halted with the introduction of imatinib and related tyrosine kinase inhibitors but restarted to address resistance to tyrosine kinase inhibitors in patients with chronic myeloid leukemia. Bedaquiline is the first molecule approved for tuberculosis (TB) in decades. 33 Drug hunters at Johnson & Johnson sought new anti-TB compounds by selecting prototypes of different chemical series and testing them for inhibition of multiple-cycle growth of Mycobacterium smegmatis. A whole-cell assay was preferred because of its ability to concurrently assess multiple targets. 33 They subsequently used resistance mutations to identify adenosine triphosphate (ATP) synthase as the site of action.
Mechanistic Starting Points
What mechanistic and chemical knowledge was used to choose the starting points for these successful drug discovery programs? The eminent drug discovers Dr. Paul Janssen and Sir James Black recognized the importance of good starting points. They emphasized the importance of starting with assays that translate to human disease.37,38 To address this question, the mechanistic understanding used as starting points for the first-in-class NMEs discovered in phenotypic assays between 1999 and 2008 was analyzed and categorized into disease biomarkers, biochemical mechanisms and pathways, molecular mechanism of action, and chemical starting points. These categories are briefly described below with the corresponding medicines. What is clear from the analysis is that in most cases, some level of mechanistic understanding was used to identify the assay format as well as the compounds used for the screens. The corresponding references are in Table 1 .
Biomarkers
A biomarker is a measurable characteristic that reflects the severity or presence of some disease state. More generally, a biomarker is anything that can be used as an indicator of a particular disease state or some other physiological state of an organism. Validated biomarkers, while reflective of disease, do not infer details of molecular mechanism. Medicines that were identified using biomarkers included ezetimibe by measuring cholesterol-lowering in animals, nateglinide by screening in a diabetic animal model, pemirolast by measuring anaphylaxis mediated by homologous IgE or IgG, rufinamide in animal models of epilepsy, and retapamulin by reassessment of early antibiotics for activity against resistant organisms. A common feature of these was a previous mechanistic understanding of the role of the biomarkers in disease. In this respect, these approaches were agnostic toward a specific mechanism but not the therapeutic indication.
Biochemical mechanism and pathways
For many diseases, medical research has identified potential pathways and biochemical mechanisms that may contribute to disease. This knowledge can be used to design assays to screen for compounds that modulate these pathways or mechanisms. These assays may be in animals or cells. The mechanistic knowledge focuses the screening to specific biochemical systems but does not predetermine the specific targets or MMOAs that will be most effective. This category included azacitidine and nelarabine, nucleoside analogues for cancer; caspofungin, a glucan biosynthesis inhibitor; cilostazol, an inhibitor of platelet aggregation; cinacalcet, a calcium agonist; miglustat, a glycolipid biosynthesis inhibitor; nitisinone, an herbicide in which new mechanistic understanding led to its evaluation for type I tyrosinemia; daptomycin, a natural product with a unique antimicrobial mechanism of action; and sirolimus, an antimicrobial with anti-inflammatory properties. With these medicines, an understanding of the biology enabled specific assays to be established and hypotheses related to disease to be tested.
It is of interest to highlight that some of the molecules were originally intended for a different indication, but once their mode of action was identified, a new medical use was tested. Miglustat originally was developed as an α glucosidase inhibitor for use as an antiviral agent. 25 Nitisinone was originally discovered as an herbicide, but early mechanism-of-action studies in rats revealed that the compound caused a marked increase in plasma tyrosine concentrations. At the same time the mechanistic studies were occurring, routine toxicity studies revealed that the drug caused corneal lesions, resembling lesions seen when rats are fed a diet high in tyrosine. This led to the conclusion that the lesions were a consequence of marked and sustained tyrosinemia. 7 Subsequently, nitisinone was evaluated in patients with type I tyrosinemia. Azacitidine, originally developed as a cytotoxic agent, was subsequently discovered to be a powerful inhibitor of DNA methylation that reduced expression and differentiation in cultured cells. 13
Molecular mechanisms of action
The assays were designed to capture MMOAs that would lead to an increased therapeutic index. The medicines included fulvestrant, a full antagonist of the estrogen receptor that achieves full antagonism via fulvestrant-induced degradation of the receptor, and the partial agonists aripiprazole (D2 receptor) and varenicline (nicotinic acetylcholine receptor). The desired molecular properties of these molecules could not be identified in a target-specific, isolated biochemical assay. With these medicines, the phenotypic assays enabled the identification of molecules that interacted in a receptor-dependent manner with the physiological system to provide an improved therapeutic index. For example, the discovery of varenicline began with the hypothesis that a partial agonist would give an acceptable dependence profile for smoking cessation. It was hypothesized that an effective agent would, through its intrinsic partial activation of the α4/β2 nicotinic acetylcholine receptor, elicit a moderate and sustained increase in mesolimbic dopamine levels, counteracting the low dopamine levels encountered in the absence of nicotine during smoking cessation attempts. At the onset of the program, a natural compound from plants known to have partial agonist activity at the α4/β2 nicotinic acetylcholine receptor, (–)-cytisine, was chosen as the chemical starting point. 27
Chemical starting points
In the above examples, the mechanistic knowledge used for the phenotypic assays was primarily biological. In addition to biological knowledge, an understanding that specific compounds were associated with a specific biological phenotype/response contributed to the chemical starting points and discovery of a number of medicines. Not surprisingly, both Janssen and Black preferred to start a discovery program with a biological active molecule.37,38 In these approaches, discovery was focused on chemical starting points with little or no mechanistic understanding. Included in this category are docosanol, for which previous studies had demonstrated antiviral activities of saturated and unsaturated alcohols of various lengths; zonisamide, a 3-substituted 1,2-benzisoxazole evaluated in anticonvulsant animal models; nitazoxanide, a nitrothiazole antimicrobial; linezolid, an oxazolidinone active against multiply-resistant Gram-positive pathogens; ziconotide, a peptide isolated from the toxins of cone snails; levetiracetam, which was evaluated in broad range of animal models of epilepsy; ranolazine, which was evaluated in animal models of cardiovascular (CV) diseases; and daptomycin, a natural product with a unique antimicrobial mechanism of action.
Serendipity
One of the interesting features of phenotypic screening is the potential for serendipity. The serendipitous discovery of vorinostat began with the observation of an effect of high DMSO concentrations on cellular differentiation. 22 Analogues were evaluated and chemical modifications eventually led to a more active analogue, suberanilohydroxamic acid (SAHA). SAHA was synthesized to test the possibility that a hydrophobic phenyl group at one end of the molecule might enhance its activity. The structure of SAHA was related to that of trichostatin A (TSA), a natural product that is also an inhibitor of histone deacetylase activity. Clinical studies occurred long before the mechanism of action identified.
Although there were success stories from serendipitous discoveries, including vorinostat, the number of medicines discovered by this approach was low. Most first-in-class medicines discovered with a phenotypic approach used chemical, biological, and/or mechanistic knowledge to focus the discovery strategy.
Identification of the Mechanism of Action
A challenge for phenotypic discoveries is to identify the mechanism of action. This brings up the questions of “how much mechanistic information is required?” and “how can it be obtained in a relevant time frame for an ongoing project?” It is difficult to accept the risk of moving a compound into development without some understanding of mechanism to help evaluate dose-response relationships to select clinical doses, predict target toxicity, and distinguish target versus off-target toxicity. Fortunately, many technologies can assist with identification of a new mechanism, including biochemical fractional isolation of activity, affinity purification, and transcriptional profiling. 39 This problem can also provide an opportunity to use new molecular technologies of chemical biology, proteomics, and network biology. The discovery of ezetimibe (Zetia) provides an example of a medicine whose predecessor was discovered in a phenotypic assay, and the target was subsequently identified using a genetic approach as the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1). 40 A more recent example by Chung and coworkers 41 from GlaxoSmithKline demonstrated the use of chemoproteomics to identify BET bromodomains as the target for inhibitors identified in phenotypic assays. A number of new methods and algorithms are being developed to identify mechanisms of action.42 –44
Contribution of Mechanistic Understanding
In drug discovery, the preferred scenario has been that molecular mechanisms associated with disease are represented by targets, and quantitation of target modulation facilitates a more rational development. However, the mechanistic details to enable this approach are not always available, validated, or sufficient for the specific medical need. It is currently unrealistic to assume that we know the exact molecular and mechanistic details of complex human diseases. The advantage of phenotypic strategies is that the exact molecular details do not have to be known.
What is clear from the analysis above is that in most cases, some level of mechanistic understanding was used to identify the assay format as well as the compounds used for the screens. The mechanistic understanding was used to focus the empirical evaluation in the phenotypic assays. One way to look at this is that starting points for drug discovery can range from an agnostic, serendipitous approach to a rational, target-based approach. The choice is based on the level of mechanistic understanding that is available to identify the assays and chemicals to be used as starting points. On the phenotypic side, there are animal and cellular assays using biomarkers, as well as the more knowledge-dependent biochemical pathways approaches. More detailed molecular mechanistic understanding allows for more success with target-based approaches. One conclusion is that a jump from a phenotypic to a rational, target-based approach without the appropriate mechanistic understanding can compromise success. Ideally, at all levels, an appropriate balance between empirical and knowledge-based approaches is warranted.
There are a number of obstacles to realizing the full value that comes from effectively reintegrating the results from phenotypic assays into the current target-focused drug discovery culture. Great progress has been made in addressing the quality of the assays and the challenges for medicinal chemistry optimization of an unknown interaction without molecular descriptors. Further efforts to understand the predictability/translation of the assays to human disease and the challenges of clinical development for a molecule with a limited understanding of the mechanism of action will lead to a greater realization of the value of phenotypic drug discovery and ultimately increase the chance for success.
Footnotes
Acknowledgements
The author acknowledges the useful comments of the editors and SLAS for providing the opportunity to publish this work.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.