
Abstract
Proteolysis targeting chimeras (PROTACs) are heterobifunctional compounds that recruit the E3 ubiquitin ligase machinery to proteins of interest, resulting in their ubiquitination and subsequent proteasomal degradation. Targeted protein degradation has generated considerable interest in drug discovery because inhibition of one particular function of a protein often does not deliver the therapeutic efficacy that results from whole-protein depletion. However, the physicochemistry and intrinsically complex pharmacology of PROTACs present challenges, particularly for the development of orally bioavailable drugs. Here we describe the application of a translational pharmacology framework (called the four pillars) to expedite PROTAC development by informing pharmacokinetic–pharmacodynamic (PKPD) understanding and helping elucidate structure–activity relationships. Experimental methods are reviewed that help illuminate exposure of the drug or probe at the site of action (pillar 1) and engagement of its target(s) (pillar 2) that drive functional pharmacological effects (pillar 3) resulting in modulation of a relevant phenotype (pillar 4). We hope the guidance will be useful to those developing targeted protein degraders and help establish PROTAC molecules as robust target validation chemical probes.
Introduction
Confirmation bias is a human tendency that negatively influences drug discovery research and development.1,2 Small-molecule chemical probes are often used to test a therapeutic hypothesis with the hope that they will phenocopy genetic methods of target perturbation such as RNAi and CRISPR/Cas9. Unfortunately, chemical probes are frequently used in excess of their potency against the annotated target in biological experiments in order to confirm the original premise of the involvement of that target in modulating a particular phenotype.3,4 Even the most selective small molecules are, by their very nature, ligands of more than one protein, and inappropriate use of chemical probes at high concentrations will result in the engagement of those “off-targets,” which could mislead research. Equally, the investigator needs to be confident that the therapeutic hypothesis is effectively tested and that the target is engaged sufficiently. This led to the development of guidance for the use of chemical probes in target validation experiments, termed the four pillars, 4 which was inspired by an analysis of the successes and failures of compounds in clinical trials. 5 The postulate is that a target validation experiment is more likely to be successful (i.e., deliver an unbiased assessment of a therapeutic hypothesis) if the four pillars are understood; these are (1) drug/probe exposure at the site of action; (2) engagement of the target (and off-targets); (3) functional pharmacological effects; and (4) perturbation of a disease-relevant phenotype. Importantly, it is the measurement of the interrelationship of the four pillars that allows for the most rigorous hypothesis testing, which can be conveyed as a set of questions: What is the effective concentration of the probe in the relevant biological system? Is the level of (free) drug sufficient to achieve target occupancy that is sufficient to drive the desired functional pharmacology? Is there appreciable engagement of off-targets that could influence the interpretation of results? To what extent does pharmacological modulation of the target alter the phenotype under investigation? Is the phenotype relevant to pathogenesis, and is it modulated to an extent that is considered therapeutic?
This framework is valid not only for target validation using chemical probes in cell-based experiments, but also for in vivo preclinical assessments and human clinical testing of drug candidates. These principles are the bedrock of successful translational pharmacology that not only establish the therapeutic relevance, or otherwise, of a new biological target or mechanism, but also inform pharmacokinetic–pharmacodynamic (PKPD) understanding. By answering the questions intrinsic to the four pillars, a more accurate PKPD assessment can be made that builds confidence in a clinical program by informing patient dose projections en route to successful proof-of-mechanism studies. Since attrition in phase 2 clinical trials is particularly high,6,7 where the efficacy of a new agent is tested in patients for the first time, it is extremely important that the reasons for failure are understood and whether the therapeutic hypothesis was effectively tested in an unbiased manner, thus demonstrating the importance of the four pillars guidance.
Proteolysis targeting chimeras (PROTACs) are heterobifunctional molecules that link a target protein ligand to an E3 ubiquitin ligase ligand (
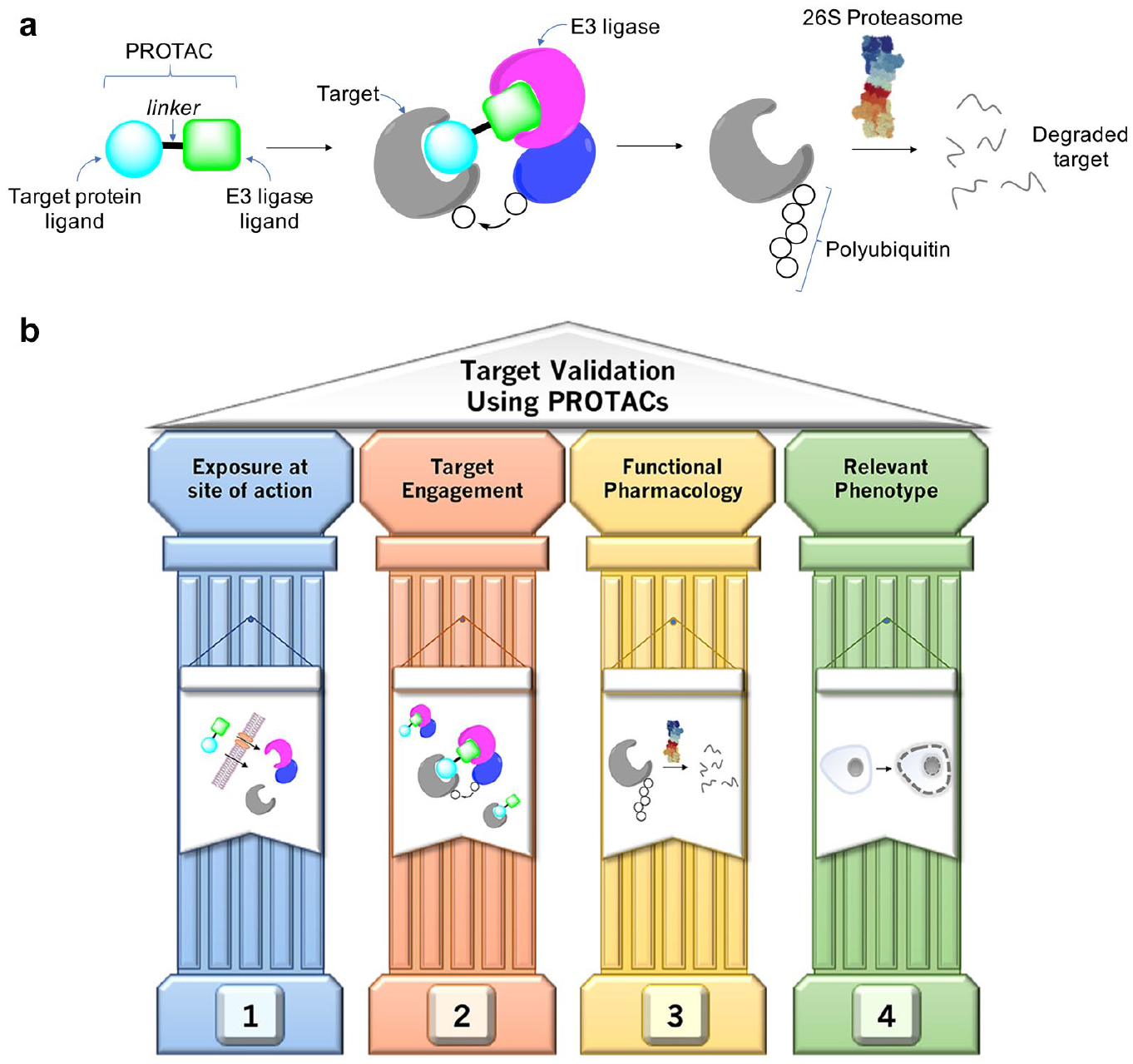
(
Despite these advances, and the apparent modular nature of PROTAC development, many of the design methodologies that could expedite the creation of such probes and clinical candidates have yet to be established. For instance, the elucidation of structure–activity relationships (SARs) is hampered by the intrinsic complexity of the pharmacological mechanism and the challenging physicochemistry associated with heterobifunctional molecules. It is worth considering these features from the perspective of the four pillars (
Once inside the cell, the physicochemistry of the molecule will influence the unbound concentration that is available to engage its targets. Nonspecific protein binding of a lipophilic PROTAC will reduce its intracellular free levels. Lipophilic compounds that also possess a basic center are often lysosomotropes and are trapped in the acidic environment of the organelle, which reduces the concentration in the cytosol (the site of action). 23 We analyzed published PROTACs 14 and discovered that 194 out of 422 (46%) possessed a basic center (i.e., protonated at physiological pH), and considering that many PROTACs are highly lipophilic, we predict that a substantial number are also lysosomotropes. High-content imaging assays have been developed 24 to evaluate lysosomotropism as a feature of small-molecule drugs that could also be used to assess the potential sequestration of PROTACs into lysosomes. Another reason to assess lysosome accumulation is the risk that lysosome dysfunction may ensue and cause toxicity, including phospholipidosis. 25
Formation of the ternary complex, and thus engagement of the target and E3 ligase by the bivalent PROTAC, is necessary for ubiquitination. The mechanism is catalytic; that is, once the target is polyubiquitinated by the E3 ligase complex and marked for degradation, the PROTAC is available to induce further heterodimerization and subsequent proteasomal degradation events.26,27 Even so, the optimization of ternary complex formation and degradation is complicated by a lack of intracellular concentration and target engagement data. Consequently, there is a considerable amount of empiricism in PROTAC development—synthetic expedience, linker length, and polarity (aliphatic carbon vs ethylene glycol chains) appear to be the prevailing design factors and the best-performing PROTACs are often empirically selected for further characterization. By delineating these complex SARs, more rational design strategies could be realized to enhance the creation of PROTACs with improved efficacy and potency. Molecular design will no doubt be enhanced in the future by the application of structural biology and computational docking models of the ternary complex in order to facilitate the optimization of selective degraders.28–33
Another challenge regarding the lack of knowledge about ternary complex occupancy is the hook effect, where high concentrations of the PROTAC cause oversaturation of the target protein and ligase that hinders heterodimerization. 34 As a result, bell-shaped dose–response curves are not unusual, and therefore understanding this phenomenon becomes a consideration for PKPD studies since high drug or probe concentrations may prevent formation of a productive ternary complex. 10 One might also expect elevated PROTAC concentrations to cause higher occupancy of off-targets that would naturally complicate the interpretation of functional pharmacology and phenotypic efficacy, as discussed above.
Ideally, proximal pillar 3 biomarker levels accurately represent drug-target occupancy (pillar 2), which itself translates to the modulation of functional phenotypic effects (pillar 4). However, this can sometimes be difficult to establish, and although some distal molecular biomarkers are technically enabled, they may not accurately report on the direct consequences of target engagement (ternary complex formation) due to the multiple inputs that are intrinsic to a responsive and controllable biological network. Additionally, the study of a proximal biomarker such as the phosphorylated substrate of a kinase may be complicated by redundancies in phosphorylation pathways and the distinct possibility that a specific phosphosite is not a pathogenic protein modification. 35 An advantage of the PROTAC modality is the ability to readily measure protein levels as a highly relevant proximal biomarker of target engagement. Arguably, there is also a higher likelihood that depletion of a network node will translate to efficacious phenotypic modulation (see “Pillar 3” section below).
Clearly, due to the challenges of developing heterobifunctional molecules described above, innovative assays are required to help understand structure–activity and PKPD relationships, and to ensure the appropriate interpretation of target validation studies when using the PROTAC modality as a chemical probe. Below we provide a perspective on the existing state of the art and detail the application of methodologies that shed light on PROTAC translational pharmacology using the four pillars framework. Additionally, recommendations are made regarding the potential utility of additional technologies to enhance the characterization and optimization of PROTACs, which could hasten their development into useful pharmacological tools and transformational medicines.
Pillar 1: Site of Action
As mentioned, there are numerous factors that prevent the PROTAC from reaching its site of action in the cytosol of the desired cell, including limited permeability, nonspecific binding to cellular biomolecules, and sequestration into subcellular compartments such as lysosomes. Readily available assays may be used to understand the potential of each of these factors, but often they serve as proxies of the specific cellular system under investigation. For instance, general membrane permeability and transporter interactions are assessed using human colon adenocarcinoma Caco-2 and Madin–Darby canine kidney (MDCK) epithelial cell lines, but it should be remembered that these assays were developed as models of intestinal permeability that facilitate high-throughput drug candidate screening. 36 Caco-2 and MDCK permeability assays are certainly useful for the development of orally bioavailable small molecules, but they may provide misleading information regarding penetration of the small molecule into the cell type being used to measure degradation efficacy. Transporters no doubt affect PROTAC absorption and biodistribution, and they are by their very nature heterogeneously expressed across different cells and subcellular organelles. 37 As an example of this, the potency of an ALK-targeted PROTAC was compromised due to active efflux from certain cell types that expressed the P-glycoprotein (P-gp) ATP-dependent exporter. 17 Unbiased functional genomic screening could be used to identify other transporters to help illuminate possible asymmetric tissue distribution in vivo and differences in degradation efficacy in different cell types and tissues. 38
Various imaging modalities could be used to assess PROTAC biodistribution, although there appears to be no reported example of this to date. Positron emission tomography (PET) imaging relies on the use of tracers containing radioactive positron emitters such as 11C and 18F. 39 Although the development of radioactive PET probes is technically challenging, recent advances in synthetic chemistry have considerably enabled the field.40,41 Stimulated Raman scattering (SRS) microscopy is used to image compound distribution in cells and tissues. 42 Interestingly, many PROTACs already contain Raman-active vibrational motifs (an alkyne, for example) 14 that would permit SRS imaging. Label-free mass spectrometry (MS) imaging methods are increasingly being used to understand drug and metabolite distribution not only at the tissue level but also at subcellular resolution as innovations continue to be made in the area. 43
Plasma protein binding (PPB) in vitro assays (using equilibrium dialysis, for example) are applied in drug discovery to help understand in vivo unbound levels.22,44 PPB is often used mistakenly as a surrogate of the nonspecific binding propensity of small molecules more generally, but this can vary considerably depending on the biological system being investigated and will lead to erroneous conclusions. For instance, phospholipids are a major sink of nonspecific drug binding and their components vary considerably across different cell types. 45 Therefore, we recommend the development of assays that support the measurement of unbound PROTAC concentrations in cells being used to measure degradation to better delineate degrader SARs. Equilibrium dialysis in combination with quantitative liquid chromatography (LC)–MS could be used to determine free drug levels in cells that should provide knowledge of intracellular PROTAC levels available to induce heterodimerization of target and E3 ligase.46–48 Determination of the unbound levels in vivo is also essential to understand PKPD relationships, as for any drug discovery program and target validation study. 4 Factors affecting in vivo free levels include PPB and nonspecific binding as discussed, metabolism, solubility, and permeability. However, it should be noted that the catalytic PROTAC mechanism may translate to a requirement for only low unbound levels at the site of action, which may be difficult to determine experimentally.
Pillar 2: Target Engagement
The catalytic PROTAC mechanism of degradation accentuates the importance of understanding the levels of target engagement that are required to achieve efficient degradation, as this will ultimately determine the clinical dose requirements. The event-driven nature of the PROTAC mechanism emphasizes the need to understand ternary complex engagement as the critical component necessary for productive degradation. It is also important to appreciate the distinction between the catalytic degradation mechanism and a classical inhibitor development paradigm where high binary receptor–ligand occupancy is desired, which is not applicable to PROTAC development because saturation of binary binding results in the hook effect as described.
Since PROTACs are able to recruit both the protein of interest (POI) and the E3 ligase, target engagement can be separated into three components: engagement of the E3 ligase by the PROTAC, engagement of the POI by the PROTAC, and formation of the ternary complex. These components can be characterized in vitro as well as in cellular systems, which are summarized below.
In Vitro Target Engagement Assays
A multitude of well-established methods can be used to assess the ability of PROTAC molecules to bind the E3 ligase or POI components in vitro using purified recombinant proteins. Examples include fluorescent probe competition assays that can be assessed using fluorescence polarization (FP) or time-resolved fluorescence energy transfer (TR-FRET), as well as probe-free methods such as surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC). While such techniques are useful to assess PROTAC binding to target or E3 ligase, these conventional assays can also be utilized to monitor the effects of binding affinity on ternary complex formation, allowing for studies of cooperativity. The protein–protein interactions (PPIs) engendered by a PROTAC molecule in a ternary complex will induce either complex stability (positive cooperativity) or instability (negative cooperativity). Cooperativity in this case is defined as the ratio of affinity in AB-C, and affinity of B-C, or similarly the ratio of affinities between A-BC and A-B, where A and C are protein partners and B is the PROTAC molecule. Biophysical methods such as ITC, SPR, or FP can be used to estimate the cooperativity of ternary complex formation, as shown in studies of CRBN and VHL-based PROTACs directed at BRD4. In an ITC experiment, VHL-EloB-EloC was titrated to a preformed complex of BRD4BD2-MZ1 and compared to its binary affinity (MZ1-BRD4BD2), leading to an estimated cooperativity of binding of ~18 to BRD4BD2. 49 The cooperativity of CRBN-based BRD4 targeting PROTAC dBET6 was measured in an FP assay by performing titrations of the PROTAC to a preformed complex of CRBN–lenalidomide–BODIPY in the presence of increasing BRD4BD1 concentrations. 28 While the VHL-based PROTAC MZ-1 indicated positive cooperativity, as measured by ITC (as well as SPR),49,50 the CRBN-based PROTAC dBET6 demonstrated negative cooperativity (<1), yet both compounds possess comparable BRD4 degradation activities. Similarly, SPR was used recently to measure the cooperativity of binding of a CRBN-based BTK kinase PROTAC, but no significant positive cooperativity was identified. 51 It is clear from these studies that while certain levels of ternary complex formation are necessary for successful degradation, the rules governing cooperativity appear to be specific to the protein–ligase pairs studied and are perhaps difficult to generalize and thus leverage in an impactful manner at this current time. However, it was concluded in the BTK degrader study that the significant improvement in potency for PROTACs possessing long linkers relative to short ones was due to the relief of negative cooperativity. 51
Cellular Engagement Assays
Measurement of E3 ligase and POI engagement in cells can be achieved by methods such as the cellular thermal shift assay (CETSA) and luciferase-based NanoBret systems. CETSA assays rely on a principle that upon an increase of temperature, proteins unfold and aggregate.52,53 Aggregated proteins are removed after cell lysis by centrifugation, and the resulting soluble protein is quantified using techniques such as Western blot or MS. 54 Small molecules capable of engaging their target often stabilize or destabilize the protein, resulting in unfolding at higher or lower temperatures, respectively, which can be described quantitatively by measuring the thermal shift described as Tm (bound ligand) – Tm (no ligand control). 53 An isothermal dose–response study yields the compound–target occupancy. The advantage of CETSA over other techniques is its ability to characterize the cellular engagement of native proteins without the need for any modifications. The outcome of the assay depends on the unfolding profile of the target proteins, which at times may be difficult to interpret and quantitatively measure.
Another commonly used assay for the assessment of target engagement is NanoBret. It relies on an energy transfer between a bioluminescence resonance energy transfer (BRET) pair: fluorophore ligand (BRET acceptor) and NanoLuc luciferase (BRET donor) activated by the addition of NanoLuc substrate furimazine. 55 Upon addition of the substrate, NanoLuc generates luminescence with a peak emission at 410 nm, which excites the acceptor fluorophore ligand. The ratio of emission to excitation generates a quantitative BRET signal. While BRET donors require bright luciferase such as NanoLuc, acceptors can be selected from a range of fluorophores with compatible excitation/emission wavelengths such as NanoBret 590 or 615 nm, 55 and the recently developed Janelia dyes. 56 Target engagement NanoBret assays observe loss of BRET signal from displacement of the fluorophore-labeled ligand by the PROTAC. The advantage of this setup is the ease of measurement in a fluorescent plate reader enabling throughput of 96- and 384-well formats. Although the BRET assay is straightforward, it is typically performed with overexpressed proteins, resulting in protein concentrations that are different than endogenous levels, which may affect the observed EC50. NanoBret cellular engagement can also use split NanoLuc partners, a high-affinity HiBit peptide, and the corresponding LgBit protein. HiBit is an 11-amino-acid peptide 57 with high affinity to the LgBit protein, which together form the functional NanoLuc luciferase. This allows for the utilization of endogenously tagged HiBit proteins by overexpressing LgBit protein to form the functional NanoLuc donor inside the cell, at concentrations limited by the HiBit-tagged protein, which is closer to a native setting. 58 BRET assays have been successfully established to measure target engagement of multiple proteins, including CRBN or VHL E3 ligase adapters, 58 but optimization of a customized assay can be nontrivial, with fluorophore tracers themselves suffering from variable cellular permeability.
In addition to NanoBret assays, a surrogate readout of cellular target engagement can be achieved by measuring the degradation of a reporter protein. An example of such an assay was reported recently that was based on BRD4BD2 and its active degrader, dBET6. 59 The compound of interest was titrated into cells stably expressing a GFP-BRD4BD2 fusion with an mCherry reporter in the presence of 100 nM dBET6. Tested compounds compete for binding to CRBN that inhibits degradation of BRD4 by dBET6, resulting in a dose–response curve enabling an assessment of CRBN engagement. These assays can be rapidly established with existing PROTAC–target pairs, but the readout is indirect, which may convolute interpretation of the results, especially if the tested PROTAC shows off-target degradation of the assay target.
Functionalized chemical probes may be used to report on the occupancy of the binding site in a protein. Clickable covalent, or photoaffinity, probes engage amino acid residues in the protein binding site in cells, which are then lysed, and reporters or enrichment handles such as fluorescent dyes or biotin are conjugated to the adducted peptide using click chemistry.60,61 The compound of interest, a PROTAC in this instance, will bind the protein (target/ligase) and prevent labeling by the functional probe, which can be analyzed using Western blot or MS proteomics. We are not aware of this approach being applied to PROTAC occupancy determination, but chemoproteomic screening technologies have been used to identify novel ligandable E3 ligases that led to the development of novel degraders,62–64 suggesting that there is potential to convert such probes into engagement biomarkers.
In Vitro Ternary Complex Formation Assays
The ability of a PROTAC molecule to form a ternary complex is a prerequisite for successful degradation, which can be measured using a variety of assay formats. One method quantifies the ternary complex directly using TR-FRET proximity-based assays, which further enable kinetics measurements. In TR-FRET dimerization, one of the protein components is tagged with a fluorophore, such as BODIPY/Alexa647 (acceptor), and the other with terbium/europium (donor). Titration of the PROTAC molecule leads to formation of a ternary complex that is measured via changes in the ratio of the acceptor/donor signal. Reagent preparation typically involves labeling one of the components with a fluorophore, which can be performed directly on the protein with cysteine or amine reactive dyes or indirectly, through the use of protein affinity tags as exemplified by the SpyTag-SpyCatcher system. 65 SpyCatcher is a small (12.3 kDa) protein that covalently binds SpyTag (13 amino acids) in a spontaneous reaction. Importantly, the SpyCatcher protein lacks cysteine residues, but a single mutation (S50C) enables specific labeling with a cysteine reactive dye. 28 Fluorophore-labeled SpyCatcherS50C can in turn be used to label proteins tagged with SpyTag. Direct chemical bioconjugation of proteins with fluorophores could label residues that interfere with the formation of PPIs required for effective ternary complex formation, and therefore indirect, but highly specific, labeling methods provide an advantage. A common strategy employs biotinylated protein and terbium-labeled streptavidin, but alternatives using glutathione S-transferase or His-tags are also readily available. 66 TR-FRET dimerization assays are readily scalable, with a high signal-to-noise ratio, but as for all light-based detection methods, they may suffer from artifacts such as autofluorescence or protein aggregation at high compound concentrations.
Cellular Ternary Complex Formation Assays
Direct assessment of ternary complex formation in cells requires a highly controllable and quantitative assay because the observed functional dimerization is often a transient event due to subsequent degradation of the POI. Deconvolution of ternary complex formation and degradation effects can be controlled, albeit imperfectly, with the addition of specific inhibitors of the ubiquitin proteasome system (UPS). Complications arising from the effects of protein degradation are especially important for PROTACs that possess rapid degradation kinetics, where treatment-to-measurement time is critical and initial formation of the ternary complex is easily missed. For compounds where the formation of the ternary complex is slower than the measurement time, the complex can be readily measured, which may also provide an indirect readout of target degradation kinetics.
Quantitative ternary complex formation assays include the recently developed NanoBret (described above) or NanoBit systems. 67 NanoBit is a split NanoLuc system, consisting of Large Bit (LgBit), a 18 kDa protein, and an 11-amino-acid peptide called Small Bit (SmBit). Differentially tagged E3 ligase and target proteins tagged with LgBit and SmBit are brought into proximity following addition of the PROTAC, which reconstitutes the active NanoLuc luciferase. The affinity of SmBit to LgBit is in the range of ~190 µM, resulting in minimal residual interactions between the tagged proteins. 67 The location of the tags, on either the N- or C-terminus of proteins, has to be empirically determined, which is a limitation of the system.
In the NanoBret dimerization assay, one of the protein partners is expressed as the NanoLuc fusion, and the other often as a HaloTag fusion. The HaloTag protein can be covalently labeled with a fluorophore–HaloTag ligand that forms the acceptor in the pair. A BRET signal is then observed upon addition of the NanoLuc substrate. The location of the NanoBret tags has to be empirically determined because there is a strict distance constraint of the NanoBit system. NanoBret assays were successfully used to monitor dimerization kinetics between BRD4 and E3 ligase CRL2VHL induced by MZ-1 or CRL4CRBN induced by dBET1. 58 The assay system has also been used to follow other steps in the UPS, such as ubiquitination kinetics of the substrate or engagement with the proteasome. 58 NanoBret assays are frequently performed as ectopic overexpressions of individual components in cells, and therefore cellular concentration of the E3 ligase and POI are far from native.
Pillar 3: Functional Pharmacology
In addition to understanding permeability, distribution, and target occupancy, PROTACs should be assessed for their functional pharmacological effects, that is, target ubiquitination and degradation in cells. In particular, although PROTACs may engage their targets and effectively induce dimerization, the resulting ternary complex may not result in ubiquitination and degradation potentially due to differences in the orientation of the recruited E3 ligase. 29 This emphasizes the need to understand the quantitative relationship between pillars 2 and 3. Protein degradation as a proximal functional readout of target engagement lends itself well to established methods of detection of protein levels such as Western blot and capillary-based immunoassays. 68 More recent techniques utilize genome editing such as HiBit assays, where the readout is luminescence, or fluorescence that leverages the exogenous expression of a fluorescent target protein fusion. HiBit relies on the split NanoLuc luciferase system HiBit and LgBit. The high-affinity HiBit tag can be placed at the endogenous locus of a target protein using CRISPR-Cas9 to generate a HiBit cell line. Upon addition of the NanoLuc substrate and either transfection of LgBit plasmid (live-cell assay) or cell lysis and addition of the purified LgBit protein (lytic assay), the HiBit-tagged protein levels can be readily measured by luminescence detection using a plate reader. HiBit assays are relatively high throughput and provide a near-native readout on protein levels that correlates well with other methods, such as Western blots. While the method has been rapidly accepted as the gold standard in the targeted protein degradation field, the development of HiBit cell lines follows the challenges present in other CRISPR-Cas9 editing strategies, such as differences in knock-in efficiencies between cell lines, the potential for off-target editing, and difficulties in isolating single-cell clones or pools with high HiBit content. Very careful cell line development and validation by sequencing are necessary.
In addition to methods specific to desired targets, a significant advantage of targeted protein degradation is the ability to utilize MS approaches to quantify changes in the cellular proteome. MS methods are ideal for the assessment of degradation selectivity, where typically 8000–10,000 proteins can be detected in a single experiment. 69 Whole-proteome detection methods can also be parallelized up to 16-plex 70 by the use of tandem mass tag (TMT) reagents, where separate treatments are labeled with isobaric mass tags, and the detection of reporter ions fragmented off the labeled peptides in MS/MS spectrum peptide assignment in each treatment.
MS proteomics is an important tool to identify PROTAC targets where degradation and inhibition selectivity can vary considerably. These lessons are exemplified by studies using promiscuous kinase inhibitors as the PROTAC target protein functional motif. For instance, a cereblon PROTAC employing a promiscuous kinase inhibitor warhead (TAE684) was found to inhibit ~193 kinases at 1 µM, but degraded only 28 kinase proteins, as determined by proteomics.71,72 Furthermore, in some cases weak binders have been converted into potent degraders. A p38α-targeted PROTAC was shown to bind the kinase with an affinity of 11 µM but results in potent degradation of the target (DC50 = 210 nM, Dmax = 91% at 24 h), indicating the complexity of the system, where ternary complex formation and induced PPIs predominate over individual warhead affinities.
Quantitative MS proteomics can also be used in target validation and identification experiments as an indirect readout of target engagement. PROTACs were prepared previously based on a phenotypic screening hit that inhibited the heat shock factor 1 (HSF1) stress pathway. 15 The probe selectively degraded a putative transcription factor called pirin that was confirmed as the relevant target of the original hit compound. 73 Similarly, a PROTAC based on the kinase inhibitor drug sorafenib was shown to degrade PDEd using proteomics, a target that was subsequently confirmed to be engaged by sorafenib itself. 74
Pillar 4: Relevant Phenotype
It is important to distinguish the differences between the consequences of inhibition and the degradation of the target. For inhibition, only a specific function of the protein is disrupted, typically localized to a functional domain. For example, small-molecule-mediated inhibition of a kinase reduces phosphorylation levels of its substrate(s) that consequently perturbs downstream signaling cascades. Degradation, on the other hand results, in removal of the whole protein, which affects both its activity and any scaffolding or noncatalytic roles that other parts of the protein may possess. 10 This difference is especially evident in the case of proteins with multiple domains, where functional degradation can manifest itself in dramatically different manners. For example, focal adhesion kinase (FAK) possesses well-established kinase-dependent and -independent functions, and kinase inhibitors have had low success in clinical trials. A FAK PROTAC was developed that not only possessed superior activity with respect to FAK activation over an inhibitor, but also addressed the kinase-independent functions of FAK (cell migration and invasion) in the aggressive TNBC cell line MDA-MB-231. 75
Therefore, it is essential that assays delineate the additional functions of a degrader versus an inhibitor, and ideally from a therapeutic perspective, these are developed in the most disease-relevant cells. It should not be assumed that the relative contributions of inhibitory function of a PROTAC versus its degradation efficacy remain the same across cell lines and primary cells, simply because PPIs and signaling events are altered in different cellular contexts. Additionally, the expression of the ligase and target, and target protein resynthesis rate, may differ across cell types, which influences PROTAC efficacy/potency and safety profiles. 51 To the latter point, recent publications reported reduced platelet toxicity for navitoclax-based PROTACs degrading Bcl-xl, due to low expression levels of the E3 ligase cereblon or VHL in platelets.76–78
It is also important to remember that PROTACs often incorporate known potent inhibitors of a particular target, which can make the distinction between the effect of degradation and inhibition difficult. In order to address these subtle differences, the use of an appropriate degradation negative control is advised. While comparison between a PROTAC and the parental inhibitor may seem appropriate, the differences in molecular properties, such as solubility or permeability between these molecules, can lead to disproportionate effects as cellular drug levels can vary and should be interpreted with care or avoided. A more appropriate matched-pair negative control PROTAC would have similar molecular properties and serve as a better comparison. Commonly used strategies abolish binding of the PROTAC molecule to the E3 ligase through removal of a glutarimide carbonyl group 59 or N-methylation of the glutarimide 79 for CRBN or use of an inactive enantiomer of the VHL ligand.26,80,81
Conclusions
PROTACs have become an established small-molecule modality with considerable therapeutic potential. Additionally, they are increasingly being developed as chemical probes to validate new therapeutic targets and as a target identification technology for phenotypic screening. 9 However, a framework is required to help guide PROTAC development. The four pillars were inspired by observations of clinical attrition and were created to establish PKPD understanding and help elucidate multifaceted SARs. We believe the roadmap is ideally suited to advancing the PROTAC modality due to its challenging physicochemistry and complex pharmacological mechanism.
Preferably, a quantitative relationship between the pillars is established in the same cell type and new assay technologies are required to realize this goal. The perspective is not an exhaustive review of the advances in the area but, rather, highlights important technological innovations and screening strategies that facilitate the progression of this important class of compounds.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors receive research funding from Deerfield.