
Abstract
Recently, mass spectrometry (MS) has emerged as an important tool for high-throughput screening (HTS) providing a direct and label-free detection method, complementing traditional fluorescent and colorimetric methodologies. Among the various MS techniques used for HTS, matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) provides many of the characteristics required for high-throughput analyses, such as low cost, speed, and automation. However, visualization and analysis of the large datasets generated by HTS MALDI-MS can pose significant challenges, especially for multiparametric experiments. The datasets can be generated fast, and the complexity of the experimental data (e.g., screening many different sorbent phases, the sorbent mass, and the load, wash, and elution conditions) makes manual data analysis difficult. To address these challenges, a comprehensive informatics tool called MALDIViz was developed. This tool is an R-Shiny-based web application, accessible independently of the operating system and without the need to install any program locally. It has been designed to facilitate easy analysis and visualization of MALDI-MS datasets, comparison of multiplex experiments, and export of the analysis results to high-quality images.
Introduction
Miniaturized high-throughput screening (HTS) methods applied for characterization of different separation phases or biomolecules have provided a breakthrough in the bioprocess development.1,2 The applicability of HTS technology has been greatly expanded by the development of new liquid handling techniques; labware concepts, such as the 96-, 384-, and 1536-well formats; and software for rapid data analysis.3,4 Many conventional HTS assays based on labeling and optical detection 5 suffer from an inherent lack of selectivity and analytical depth. This is particularly troublesome during analysis of complex biological samples, as cross-reactivity and matrix effects can lead to false results. 6 Another drawback is that the development of assay-specific labeled probes or antibodies can add cost and time to the HTS process. Therefore, the label-free and direct detection provided by mass spectrometry (MS) has become an attractive method for the identification and characterization of biomolecules. 6 Surface-based MS techniques, such as desorption electrospray ionization (DESI) and matrix-assisted laser desorption/ionization (MALDI),7,8 represent particularly attractive methods due to the combination of a label-free and relatively interference-resistant readout. With a throughput (around 1 sample/s), these techniques can also begin to compete with the speed of traditional fluorescence-based techniques.8,9 In addition, these MS techniques use a very small sample volume (micro- to nanoliters), thus providing both cost-efficient analysis and the option to analyze more sample replicates.
A major bottleneck in MS-based HTS lies in the analysis of the large amounts of raw data generated. The ability to perform fast data analysis and trend identification is key for cost reduction, decision making, instrument usage, and design of further experiments. Preferably, the data analysis should be fast enough to allow for the adaption of experimental parameters during the actual HTS campaign.
Any software used for the analysis of multiparametric MS datasets should allow the user to easily organize, simplify, visualize, and analyze the data, while retaining the relationships among the different samples/parameters. 10 While there are many software options for analysis of raw MS data, there are fewer alternatives allowing for differential analysis. There are some devoted vendor-specific software tools for analysis of HTS MS data, for example, the RapidFire Integrator software; alternatively, more general tools, such as R, 11 SPSS, and Matlab 12 packages, can be used. In the case of MS-based proteomic datasets, there are a great variety of open-source libraries and frameworks available, such as OpenMS, Proteowizard, MsInspect, and Bioconductor. 13 The statistical analysis is of utmost importance for HTS data, and here DanteR 14 and InfernoRDN 15 can provide a user-friendly graphical interface to the R framework for normalization, ANOVA, and various other multivariate analysis techniques. Perseus, 16 a part of the MaxQuant package, also provides statistical analysis tools that are similar to those of DanteR. However, most of these tools are streamlined for data generated by ESI liquid chromatography (LC)–MS or require a certain knowledge of bioinformatics and sometimes even programming skills.
Outside of bacteria typing software, there are a lack of software tools that cover HTS-scale MALDI data visualization and analysis. In this article, we present MALDIViz, a comprehensive informatics tool for MALDI HTS data that can facilitate visualization, statistical analysis, and high-quality image and data export. MALDIViz is an R-Shiny-based web application, accessible independently of the operating system and without the need to install the program locally. The functionality of MALDIViz application is demonstrated by analysis of in-house-generated screening data from an investigation of different solid-phase extraction (SPE) materials 17 and a publicly available cancer dataset. 18 The MALDIViz application is accessible through https://jkkishore85.shinyapps.io/maldiviz/.
Materials and Methods
Built-In Analysis Functions
MALDIViz incorporates algorithms for multidimensional visualization, statistical analysis, and creation of publication-quality images. It has been implemented entirely in R 11 (R version 3.2.3) and developed using Rstudio and the Shiny framework. Rstudio is an open-source interface for the development of R applications, and Shiny is a package that allows the creation of web applications directly from R.
MALDIViz uses several R packages internally. The shinydashboard package has been crucial for the design of the MALDIViz application. This package provides a theme on top of Shiny, allowing the creation of visually attractive dashboards. Shinydashboard was used to create interface items, such as the side menu, and facilitates the user’s navigation through the different tabs. Other R packages, data.table, 19 reshape, 20 and plyr, 21 are used for basic data manipulation, for example, filtering and grouping. The rgl, 22 plotly, 23 and ggplot2 24 packages are used for the generation of plots. The d3heatmap and pheatmap 25 packages are used for drawing heatmaps. The MALDIquant 26 package was implemented for MALDI data preprocessing, and the pcamethod 27 package for principal component analysis (PCA) calculations.
Datasets
Dataset 1: Solid-Phase Extraction Screening Dataset
This dataset comes from a screening experiment with different molecularly imprinted SPE phases developed for the selective enrichment of a proteotypic peptide from the protein Pro-GRP, a biomarker used for small-cell lung cancer diagnosis. Briefly, six different SPE phases in duplicate were examined using four different SPE conditions using a 384-plate-based micro-SPE platform. Thereafter, the collected eluents were analyzed in triplicate by MALDI-MS. 17
Dataset 2: Cancer Dataset
This is a publicly available dataset from a MALDI-based sample profiling publication using gold nanoparticles in order to separate the proteins and peptides in human serum. The protocol described in this work divides each sample into two subsamples: pellet and supernatant. The MALDI spectra of both subsamples are grouped by their corresponding conditions using three-dimensional (3D) PCA. The dataset is composed of MALDI spectra from five patients with lymphoma, five patients with myeloma, and two healthy donors. 18
Data Visualization and Analysis Pipeline
In MALDIViz, users can browse through the visualization and analysis steps by going to each of the tabs in the left side panel. All tabs work in a similar fashion; for example, changing the settings in the left panel will automatically renew the image or table on the right. When moving from one tab to another, all settings are saved automatically.
The menu has been divided into four basic groups, each containing the operations the program can perform: “Data Import,” “Spectra View,” “Analysis – Comparison,” and “Analysis – Classification” ( Fig. 1 ).
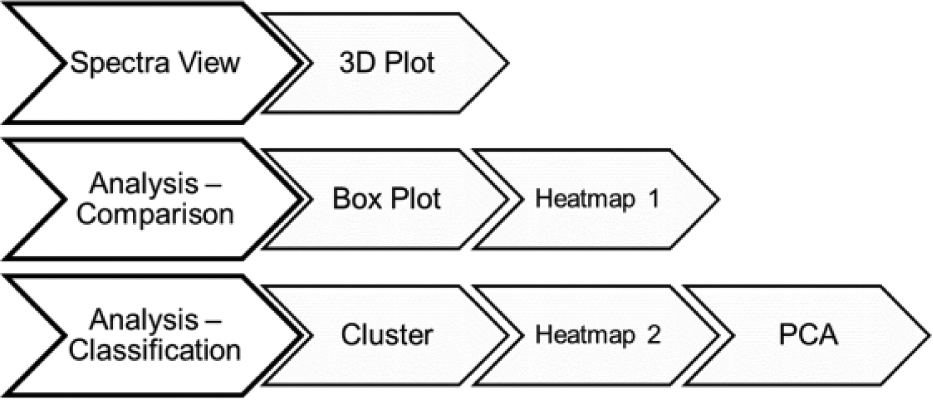
Outline of the operations that MALDIViz can perform.
Data Import
Users can directly load datasets from any file that stores tabular data, including flat files (either CSV or tab-delimited text files) and Microsoft Excel files (.csv), or similar formats. The functionality can be tested by loading the predefined datasets, available in the online app. Input data for analysis can be any data table (cross-tab) containing columns of mass and intensities, with additional columns corresponding to informational metadata from each experiment, such as type of separation phase, buffer condition, replicate details, or clinical outcomes. In an interactive analysis, the user might want to modify the sample groups, for instance, to include or exclude certain samples. To enable an efficient sample grouping, the user can change sample selection based on supplied metadata. This strategy is easy to use and particularly suited for batch processing. In the present version of MALDIViz, up to four columns of metadata can be added by the user. A screenshot from the web application showing a typical dataset loaded is displayed in Figure 2 .
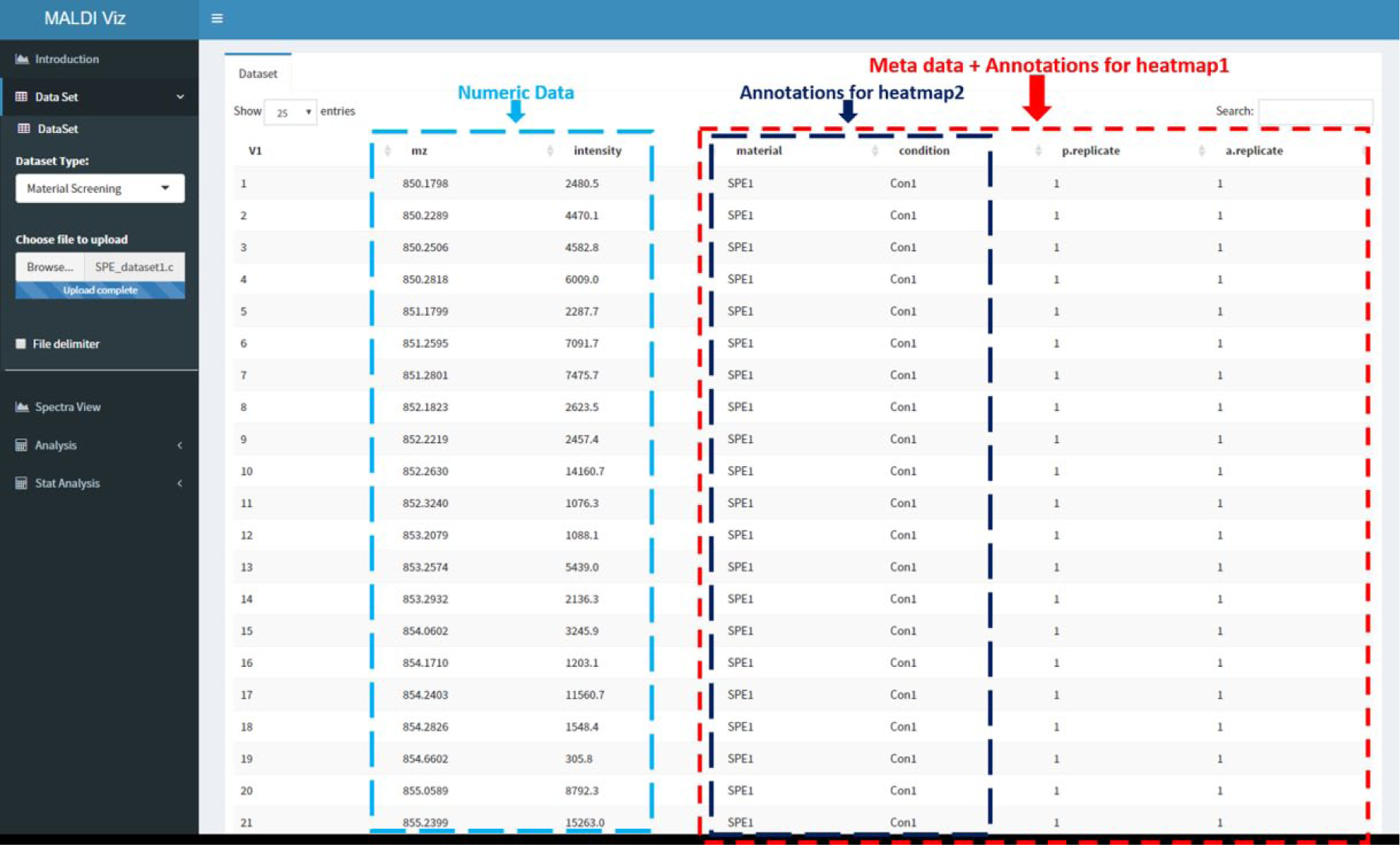
Screenshot from the MALDIViz app showing a typical dataset loaded.
Spectra View
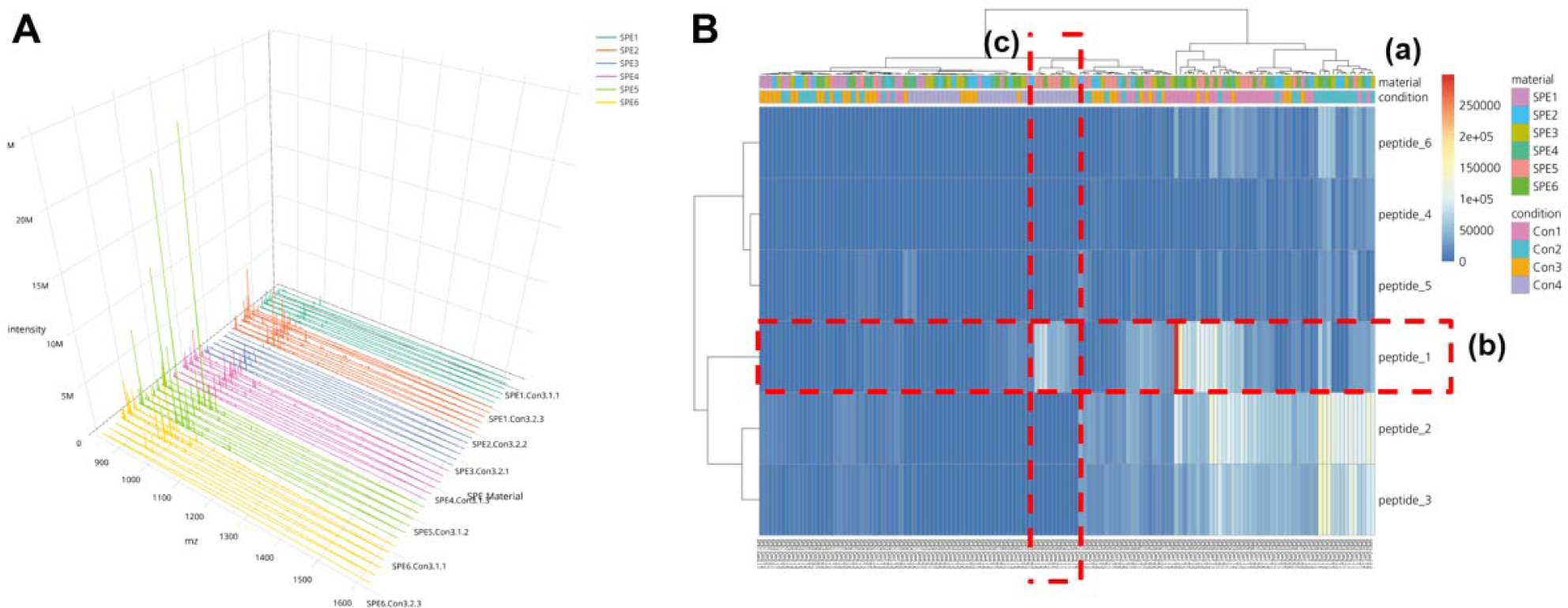
To compare multiple mass spectra from different samples, two-dimensional (2D) visualization is efficient in showing many data points at the same time. However, it is sometimes difficult to visualize the data trends when there are large overlaps. Spectra View provides a 3D plot, as an optional way to visualize and compare large datasets. In the 3D plot, in the horizontal axis, x represents m/z; in the vertical axis, y represents intensity; and in the depth axis, z represents different samples. This type of 3D plot is very useful for finding features and patterns across the experiment in one quick glance instead of looking at all the individual spectrum. The user can inspect data values such as m/z, intensity, and sample details by moving the cursor along the spectrum. Moving, scaling, and zooming functions are also made as easy and user-friendly as possible, and most manipulations are accessible by mouse. To show the 3D plot from a variable viewpoint, the user can zoom and rotate the view around all axes. Figure 3A shows an example of the 3D plot provided by MALDIViz.
(
Analysis – Comparison
The “Analysis – Comparison” window allows for comparison of the experimental data using grouping based on user-supplied values in the metadata. Significant differences between groups can be visualized using the boxplot and heatmap functions. The boxplot provides a visual interpretation of the differences between groups and a measure of the within-group variation. For example, SPE material screening data can be plotted with a grouping based on buffer condition to examine any significant differences due to varying buffer conditions. Heatmap1 is generated using pheatmap and d3heatmap R packages. Similar to the boxplot, the grouping of the data is accomplished based on user-supplied metadata, as in Figure 3B . In the heatmap, the vertical axis represents the samples and the horizontal axis represents the different masses and the colors’ corresponding to intensities. Annotations based on the metadata can also be added on top of the heatmap, to easily identify which groups are separated. Optionally, rows and/or columns in the heatmap can be clustered based on user-defined distance and linkage methods.
Analysis – Classification
The analysis classification view offers more powerful, but still easy-to-use multivariate statistical analysis tools for data exploration and validation.
Principal Component Analysis
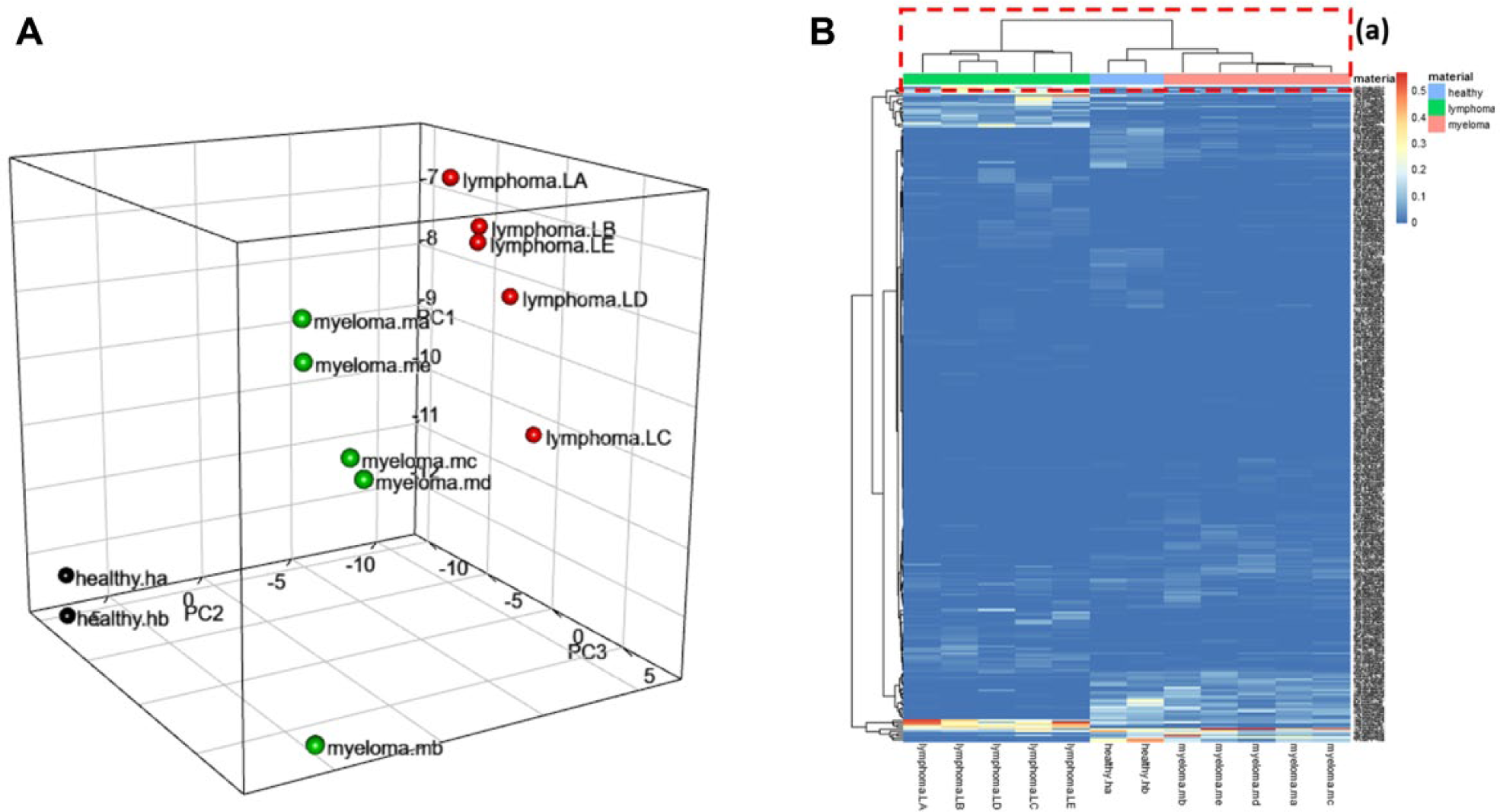
MALDIViz can provide user-selectable options for generating a 3D PCA plot ( Fig. 4A ), for classification and identification of major sources of variation in the data. PCA is an unsupervised feature selection technique used for data compression, information extraction, and preliminary visualization of observations or samples. The ultimate goal of PCA is to reduce the dimensionality of a multivariate dataset while retaining the variation in the dataset.10,28 By reducing the dimensionality, each sample (spectrum) can represented by a point in a 2D or 3D coordinate system (score plot), in which spectra with similar variation characteristics can be clustered together and the differences between sample groups can be readily visualized.
(
PCA creates new variables, the principal components (PCs), which are linear combinations of the original variables (i.e., m/z values). The first PC (PC1) is the direction along which the samples show the largest variation. The second PC (PC2) is the direction uncorrelated to the first component along which the samples show the largest variation. The first three of the orthogonal and linearly independent new coordinates (PC1, PC2, and PC3) are then used to visualize the data as dots in a 3D space. 29
It can be estimated how much each of the original variables (e.g., m/z values) contributes to each of the new PCs. These values are called loadings. The loading plots of the PCA provide information regarding the contribution of each m/z value to the variance covered by each respective PC. MALDIViz also offers two types of data preprocessing: mean centering and variance scaling prior to PCA. 30 By mean centering, an average data spectrum is calculated and subtracted from each spectrum of the dataset. Without mean centering, the calculated PC will be the average of the dataset and will not describe the variation in spectral content. Variance scaling can be used to ensure that all variables contribute equally during the PCA calculation.
Cluster Analysis
The cluster analysis aims to identify similarities or differences between the different groups in a dataset. There are several ways to define dissimilarity (or distance); 31 typical clustering methods include hierarchical clustering, k-means, neural network, and SPADE. MALDIViz provides hierarchical clustering analysis (HCA). HCA generates clusters by simply pairing individuals or subclusters that are the smallest distance apart in space, with the original parameters as the coordinates (in our case, m/z values). 32 MALDIViz allows the user to choose between different distance measurement algorithms, such as Euclidean, maximum, Manhattan, Canberra, binary, or Minkowski distance, and select different linkage criteria functions, such as complete, single, average, ward.D, ward.D2, McQuitty, median, or centroid. 31 The results of the hierarchical clustering are presented in a dendrogram. A dendrogram is a tree graph whose terminal vertices (“leaves”) correspond to the objects classified. The dendrogram is usually drawn upside down: the leaves are at the bottom and the root is on top. The y axis indicates the “distance” between individual observations and groups of observations.
Heatmap2
Heatmap2 works similarly to heatmap1, but it is plotted with hierarchical clustering and annotation options available for columns and/or rows based on the user metadata. Another important distinction is that heatmap2 plots all m/z values in the dataset, while heatmap1 plots a heatmap only containing user-defined m/z values.
Interactivity of the Plots
The interactive mode is available for all plots (3D plot, PCA plot, and heatmap), which allows the user to click, zoom, and hover over specific areas of the plot for additional information.
File Download
The plots and data generated by the MALDIViz application can be saved to the local file system as .eps, .pdf, and .csv files by clicking on the “Download” button located above each plot.
Results and Discussion
MALDIViz is an interactive web application that allows users to visualize and analyze MALDI HTS data and explore the results. To demonstrate the application, two different datasets, a SPE screening dataset and a cancer dataset, were used.
Case 1
The first case is an analysis of a dataset from a SPE screening experiment with MALDIViz. The aim of this screening experiment was to find the best SPE material among some newly developed molecularly imprinted polymers, and also identify a good SPE condition for selective enrichment of a proteotypic peptide from the protein Pro-GRP. Briefly, six different SPE materials in duplicate were examined using four different SPE conditions in a micro-SPE platform. 17 Without dedicated analysis software such as MALDIViz, a user will have to compare these mass spectra manually, which is both tedious and very risky from an analytical perspective. The visualization methods, in combination with user-defined sample grouping by coloring, provided a fast and convenient means to identify the experimental trends. From the Spectra View 3D plot, it is evident that at condition 3, SPE material 5 (light green) has fewer background peaks than SPE materials 1 (dark green), 2 (red), 3 (blue), 4 (pink), and 6 (yellow) ( Fig. 3A ). While this type of quick visual observation of the intensity distribution across the groups can be used to find features and patterns across the experiment, it is important to note that the MALDI signal intensities are not ideal for making analytical conclusions, due to ion suppression and hot spots. 33 Furthermore, there might be experimental differences affecting the intensity, for example, capacity differences in SPE materials.
To further assess experimental quality, the peptide intensity profile of the analyte peak (i.e., peptide 1) and the background peaks (i.e., peptides 2–6) resulting from analysis of the six SPE materials for four different conditions was plotted as a heatmap ( Fig. 3B ). In this heatmap, the vertical axis represents the samples, the horizontal axis represents the different peptides, and the colors correspond to peptide signal intensities. The highest intensities are easy to find in this type of plot, and the top-performing SPE material and condition is provided by the annotation available at the top of the plot. Optionally, MALDIViz hierarchical clustering can be applied to columns and/or rows. Here it is possible to choose between different clustering and linkage methods. Also, the annotation parameters can be selected based on the supplied metadata. From Figure 3B , conditions 1 and 2 were identified as having a generic SPE affinity with high recovery observed for all peptides and a low selectivity (high intensity for both target and background). While condition 4 provided two SPE materials (i.e., SPE material 5 and 6), with selective retention as the highest signal was observed for peptide 1. This significant phenomenon was also demonstrated with the boxplot analysis. 17 Summing up, MALDIViz application provided a convenient “all-in-one” data analysis platform that enables HTS of multiple different SPE materials while simultaneously identifying optimal SPE conditions.
Case 2
In the second case study, a previously published and validated dataset from a MALDI-based sample profile carried out with gold nanoparticles as a separation phase is used for demonstration. 18 The dataset comprises observed MALDI peaks after analysis of serum from five patients with lymphoma, five patients with myeloma, and two healthy donors. López-Cortés et al. 18 demonstrated that the resulting MALDI spectra could be grouped by their corresponding cancer type using 3D PCA.
Two separate classification methods, heatmap and PCA, were applied to the dataset using MALDIViz, to ensure that the previously classified results could be reproduced. For distance measurement and linkage options in the heatmap, the commonly used Euclidean algorithm with Ward’s linkage method 31 was used. The dendrogram and annotation on top of the heatmap show ( Fig. 4B ) a clear classification of the samples into three groups (i.e., lymphoma, myeloma, and healthy samples). Figure 4A shows the PCA plot generated by MALDIViz after processing the cancer dataset. The PCA classification in a 3D subspace spun by the first three PCs also resulted in three separate clusters representing lymphoma, myeloma, and healthy samples. By assigning different colors to each group (i.e., lymphoma, myeloma, and healthy), it was possible to quickly visually identify the separate groups.
In conclusion, the herein presented MALDIViz web application facilitates the analysis and visualization of HTS-generated MALDI-MS data, as well as export of the results as high-quality images for documentation or publication. The case studies provide some examples of how the visualization can facilitate identification of differences between sample groups. There is a wide range of statistical tools incorporated in MALDIViz, for example, clustering and PCA, all with a user-friendly interface designed to minimize the need for bioinformatics expertise and complicated client installations. The MALDIViz application has been and continues to be a very important tool for analysis of MALDI-MS data in our laboratory, and we now would like to share this tool with the scientific community. The application is very generic and can certainly be applied to many different analytical problems. Therefore, we also would like to encourage anyone working with MALDI-MS to try the tool; we recommend that users start with the example datasets and the quick-start tutorial (Supplementary Information). Although designed specifically for analyzing MALDI data, the MALDIViz web application can also be applied to analysis of data from microarray and other -omics data with a compatible data format.
A test server running the MALDIViz web application can be accessed freely at https://jkkishore85.shinyapps.io/maldiviz/. Efforts are also underway to add tools for quantitative analysis and missing value imputation. Feel welcome to contact us with feedback.
Footnotes
Acknowledgements
Support from the Swedish National Infrastructure for Biological Mass Spectrometry (BioMS) is gratefully acknowledged.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been performed as part of the ITN project PEPMIP (grant agreement no. 264699) supported by the Seventh Research Framework Programme of the European Commission under Marie Curie actions.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.