
Abstract
Screens using high-throughput, information-rich technologies such as microarrays, high-content screening (HCS), and next-generation sequencing (NGS) have become increasingly widespread. Compared with single-readout assays, these methods produce a more comprehensive picture of the effects of screened treatments. However, interpreting such multidimensional readouts is challenging. Univariate statistics such as t-tests and Z-factors cannot easily be applied to multidimensional profiles, leaving no obvious way to answer common screening questions such as “Is treatment X active in this assay?” and “Is treatment X different from (or equivalent to) treatment Y?” We have developed a simple, straightforward metric, the multidimensional perturbation value (mp-value), which can be used to answer these questions. Here, we demonstrate application of the mp-value to three data sets: a multiplexed gene expression screen of compounds and genomic reagents, a microarray-based gene expression screen of compounds, and an HCS compound screen. In all data sets, active treatments were successfully identified using the mp-value, and simulations and follow-up analyses supported the mp-value’s statistical and biological validity. We believe the mp-value represents a promising way to simplify the analysis of multidimensional data while taking full advantage of its richness.
Keywords
Technologies that produce multidimensional readouts for biological assays have become increasingly widespread in recent years because of their ability to capture detailed measurements at ever-increasing scales.1,2 One such technology, high-content imaging, has transformed traditional microscopy, producing dozens of numeric cytological measurements for single cells in a high-throughput fashion.3,4 The latest microarrays for gene expression interrogate more than 1 million exons, and next-generation sequencing technologies are capable of calling hundreds of billions of bases per run.5,6 By producing more than one readout for each sample input, these methods are inherently multidimensional. Synthetic multidimensional data sets are also created by combining data from disparate sources or experiments. Such data sets have been produced to create compound profiles based on kinase assay results, side effect annotations, and cytotoxicity measures in diverse cell lines.7–10
Confronted with the wealth of information in a multidimensional screening data set, it is not always clear how to proceed with the analysis in a way that can make use of the abundance of available information. Sometimes, a data set may be multidimensional simply because it is easy to collect many readouts, even when only a single readout is wanted; high-content screening (HCS) is an example, in which multiple cytological measurements can be calculated based on one stain just as easily as a single measurement. In these cases, the analysis is easily simplified by narrowing the focus to a single readout and ignoring the rest. Statistically, the problem is now univariate, and methods such as the t-test or the nonparametric Wilcoxon rank-sum test can be used to compare replicates of each treatment to the replicates for each control. However, in many situations, there is no a priori decision made to analyze a single readout from an otherwise multidimensional readout. In these situations, analytical metrics should make use of all available readouts to take full advantage of the data set.
An often-used workflow for processing multidimensional data consists of first normalizing treatments to a negative control (such as vehicle) and then applying machine-learning approaches (such as principal component analysis, support vector machines, k-means, or hierarchical clustering) to discern biologically defined groupings of the samples.11,12 Although this workflow has been used to create functional groupings of samples including cell lines, yeast deletion mutants, and tissues from drug-treated rats, it aligns imperfectly with the language of biological screening.13–15 In a screen, there are often negative and positive controls, and treatments are expected to be either active to varying degrees (similar to a positive control/dissimilar from a negative control) or inactive (similar to a negative control). Frequently, a treatment in a screen must display a measured activity beyond a certain threshold to be considered a hit and to progress for further evaluation. This threshold, which serves to remove inactive treatments, is missing from the grouping-based multidimensional data-processing workflow described above.
The normalization step common to the previously mentioned workflow makes it easy to understand comparative differences between two treatments, but it does so at some expense. Namely, the workflow does not use all of the information present in the replicates for both treatments and negative controls. Usually, replicates are summarized first by taking the means or medians of their readouts so that a single treatment profile can then be divided by a single vehicle profile. This summarization discounts variation across the replicates, losing information on the amount of noise present for each readout. In addition, in the traditional workflow, summarized negative control profiles are used only for normalization, not for direct comparison to treatment profiles. Although the use of negative controls for normalization is often seen as a necessary or logical step to reduce batch effects, some evidence suggests, counterintuitively, that it may increase them. 16 By freeing the negative control replicates from the requirement that they be used for normalization, it becomes possible to directly compare treatment replicates to negative control replicates using the same methods by which a treatment would be compared with any other treatment.
Here, we describe the multidimensional perturbation value (mp-value), a single statistic that can be applied to various types of multidimensional screening data and used to determine whether any two treatments differ from each other. The mp-value fully uses all measured readouts as well as all replicates. Calculation of the mp-value is straightforward and based on well-established statistical techniques. It lends itself easily to visualization and is similar in spirit to the well-understood p value. Here, we have used the mp-value to analyze treatments from two gene expression data sets and a high-content imaging data set and have found it to be a simple, biologically sound method for determining whether treatments are active in a given screen. We provide R code and a Web tool for calculating mp-values in order to enable easy application of this method to other data sets.
Materials and Methods
Multidimensional Perturbation Value (mp-Value) Calculation
The mp-value can be used to compare any pair of treatments: a screened treatment against vehicle control(s), a screened treatment against known positive control(s), one batch of a treatment against another batch, and so forth. To simplify the description of the mp-value calculations, the comparison of a screened treatment (“treatment”) to a negative control (“vehicle”) will be used here. A treatment different from the vehicle is considered active, whereas a treatment similar to the vehicle is considered inactive.
A general overview of the mp-value calculation can be found in Figure 1 . All calculations described in this section were performed using R, and an R function for calculating the mp-value can be found in the supplemental material. A Web server is also available for calculating mp-values (http://www.jannahutz.com/mpvalue/). A small example data set is available for download from that site and is used in the supplemental material for a more detailed, step-by-step walkthrough of how the mp-value is calculated.
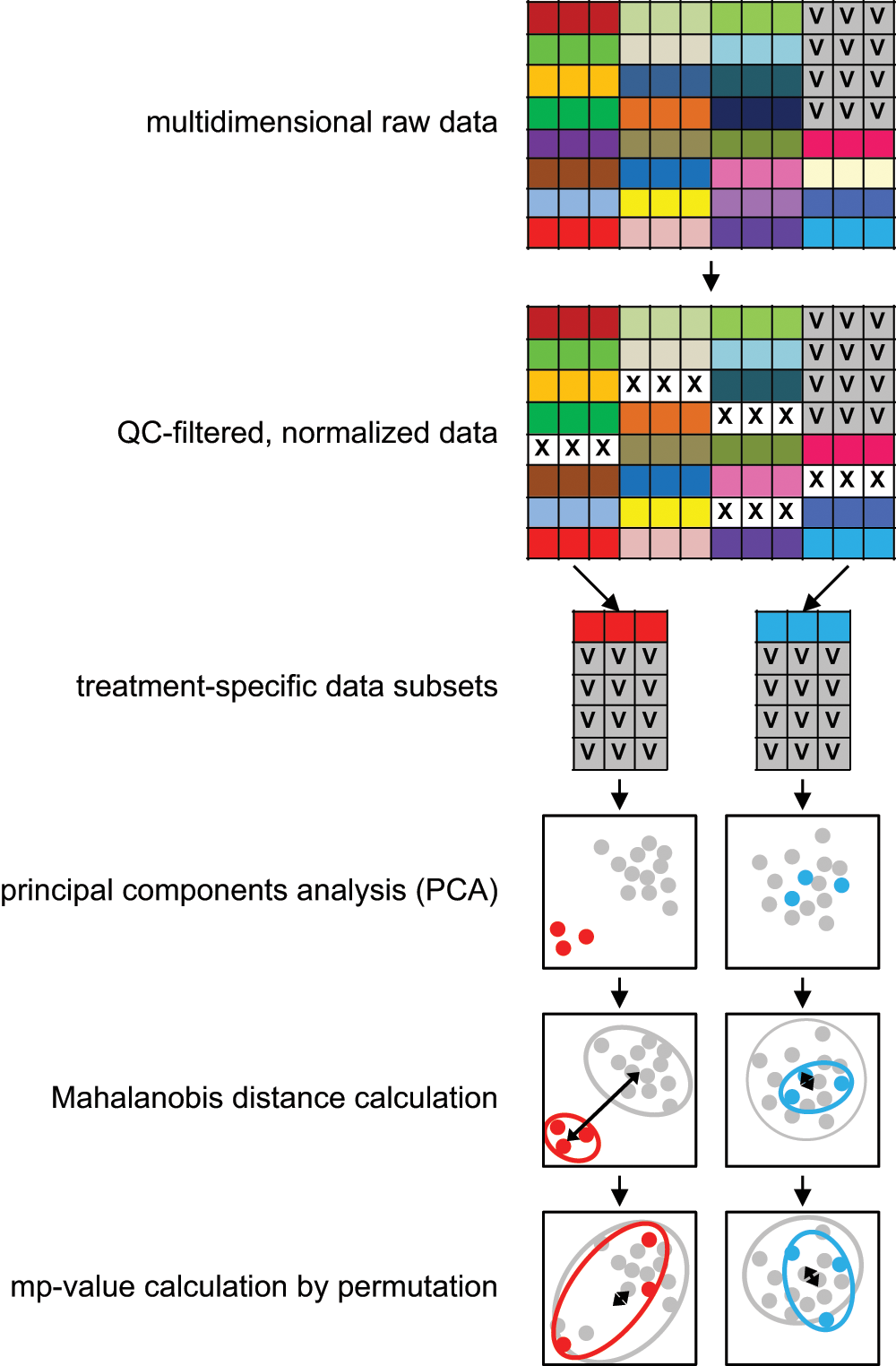
Overview of how the mp-value is calculated. Different treatments are illustrated with different colors, and each treatment here is represented in triplicate. After removing treatments that fail to pass quality control filters and performing normalization, multidimensional data subsets are created for each treatment, containing replicates for that treatment as well as vehicle replicates from the same batch. Principal component analysis (PCA) is performed on this two-treatment data set, and components are scaled by the proportion of the total variance that they explain. In this adjusted PCA space, the Mahalanobis distance is calculated between the two treatment groups. Treatment labels are permuted 1000 times (disallowing the original label configuration), and Mahalanobis distances are calculated each time. The mp-value is equivalent to the percentage of permuted Mahalanobis distances greater than the original.
Data selection and normalization
Selection of data points to use for mp-value calculation must take into consideration two properties of the mp-value. First, at least one group (treatment or vehicle) must consist of more than one replicate. Common screen designs, in which treatments are run in duplicate or triplicate and controls in higher numbers, fit this requirement. Second, a statistically significant mp-value is achieved when the multidimensional profiles of the treatment replicates are different from those of the vehicle replicates. To minimize the possibility of a significant mp-value arising from systematic experimental bias rather than biological differences, it is recommended that treatment replicates be matched with vehicle replicates from the same experimental batch. If this is not possible, raw data should be adjusted for batch effects or other sources of experimental variation before combining data from multiple batches. Any replicates failing standard quality control (QC) checks should also be removed before calculating mp-values.
These data are provided to the mpvalue R function, which next creates treatment-specific data sets. For each treatment to be evaluated, its replicates and the replicates of the vehicle control are combined to form a m-by-n data set
The example data available for download illustrate the above points. It consists of a single batch subset of normalized, QC-filtered data from the Broad Connectivity Map data set.
17
The vehicle control (DMSO) was run in six replicates in this batch, satisfying the requirement that at least one group in all treatment versus control comparisons have more than one replicate. Two example treatments, clozapine and thioridazine, are profiled in the supplemental material. For each treatment, the
Principal component analysis (PCA)
Next, PCA is performed on
To reduce the dimensionality of
Mahalanobis distance calculation
Next, the Mahalanobis distance is used to assign a value to the separation of the treatment replicates from the vehicle replicates in PCA space. The formula for the calculation of the Mahalanobis distance is shown below, in equation 1.
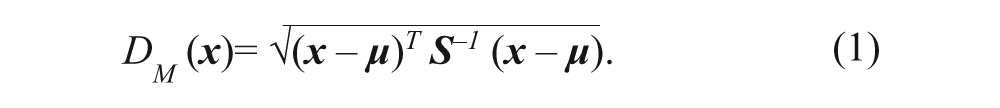
Vector
Permutation, mp-values, and significance cutoffs
To assign an mp-value (analogous to an empirical p value) to the Mahalanobis distance, a permutation approach is taken. Permutation tests are commonly used to determine statistical significance when an underlying statistical distribution is unknown and have often been used to analyze biological data. 18 Here, the null hypothesis assumes that there is no difference between the treatment replicates and the vehicle replicates. Therefore, under the null hypothesis, shuffling the replicate labels would not result in a drastically different Mahalanobis distance between the two groups. In contrast, if the treatment replicates are significantly different, the Mahalanobis distance would likely be smaller after randomly shuffling the replicate labels.
To calculate the mp-value, replicate labels for all rows in
If a treatment is run in more than one batch, mp-values can be considered separately for each batch or combined. Here, mp-values for any treatments run in multiple batches were combined using Stouffer’s method. Significance cutoffs were established using a false discovery rate of 0.1. To simplify analyses in which the negative log10 of the mp-value is taken, any mp-values equal to zero are set to 0.0001.
Simulations and Comparison to Other Statistics
Simulations of the mp-value’s performance were conducted by repeatedly generating vehicle replicates and positive and negative treatment controls by sampling readouts from a number of different normal distributions. Each treatment control was simulated in duplicate, and 16 replicates were simulated for each vehicle. One hundred readouts were simulated. All readouts were simulated in the vehicle and negative control replicates by sampling from the standard normal distribution. Varying numbers of readouts were simulated in positive treatment control replicates by sampling from other normal distributions (μ = 1, 2, or 3 with σ = 1). Any remaining readouts in positive treatment control replicates were sampled from the standard normal distribution. The mp-value was considered to be accurate if its value was statistically significant (mp-value <0.05) for a positive control treatment or it if was not significant (mp-value >0.05) for a negative control treatment.
The mp-value was compared with three other statistical methods. For the first method, each of the 100 readouts was subjected to a t-test to determine whether that particular readout was significantly different between the treatment and control replicates. A similar methodology was used for the second method, substituting a Wilcoxon rank-sum test for the parametric t-test. In both cases, the 100 resulting p values were combined using Stouffer’s method for meta-analysis, producing a single p value representing the difference between treatment and control replicates. Meta-analysis p values were considered accurate if they were significant for positive control treatments or nonsignificant for negative control treatments. The third method was k-means clustering using the default parameters for the R kmeans function (k = 2). If the k-means clustering grouped only positive control replicates in one cluster and only vehicle replicates in the other, it was considered successful. For the negative control replicates, if they were not separated into a cluster distinct from the vehicle replicates, this was also considered successful.
Experimental Data Sets
The first data set, referred to here as LMF, was generated from a medium-throughput gene expression screen of compounds, siRNAs and cDNAs. The experimental method quantifies gene expression by using flow cytometry to detect levels of fluorophore-labeled cDNA bound to probe-specific beads and has been described previously. 19 For each treatment, the same set of 100 probes was assayed, corresponding to readings for 97 unique genes. Negative controls were DMSO for compound treatments, scrambled siRNA for siRNA treatments, and an empty vector for cDNA treatments. Additional information on this data set as well as the following two data sets can be found in the supplemental material.
The second data set, referred to here as CMap, is the gene expression microarray-based Connectivity Map data set created and described by the Broad Institute. 17 A subset of the microarrays in this data set that shared experimental conditions was used here, consisting of 2674 arrays comprising 1150 unique compound treatments. Arrays were checked for quality using MDQC and normalized using ComBat.20,21
The third data set, referred to here as HCS, resulted from a screen of a collection of 3805 unique natural product compounds and has been published previously. 4 Normalization was done using median polish.
Results and Discussion
The mp-value was first assessed using data simulated to reflect a typical biological screen. Treatment samples were simulated to either have no difference (negative control treatments) from the simulated vehicle replicates or to differ from the vehicle (positive control treatments) in one or more readouts. In this simulation, the mp-value was highly accurate in calling negative controls, and it also performed well in identifying positive controls ( Fig. 2 ). It was more successful when more readouts differed between the treatment and vehicle replicates or when readouts differed by a larger margin between the two groups ( Fig. 2 ).
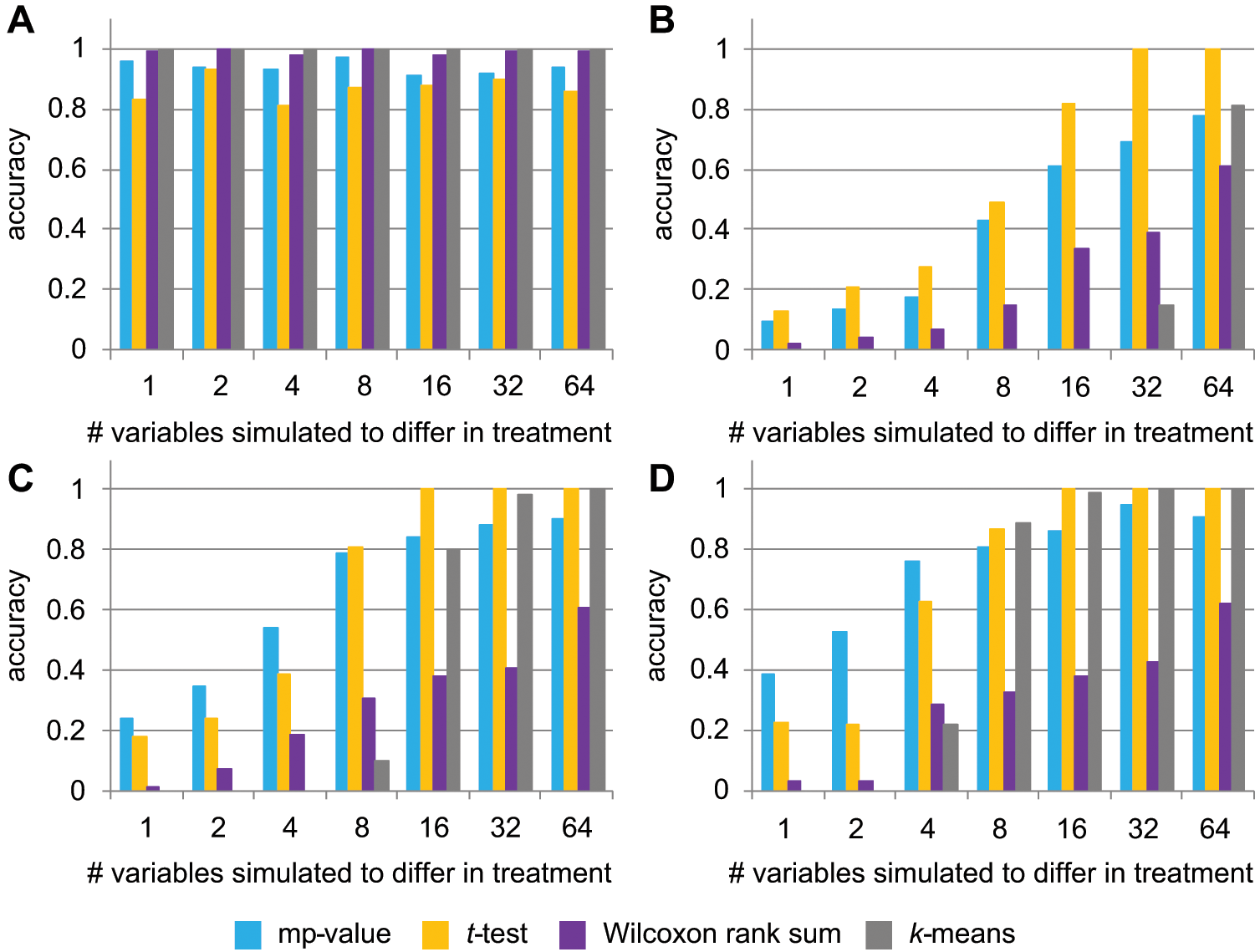
Performance on simulated data of the mp-value (blue), the t-test (orange), the Wilcoxon rank-sum test (purple), and k-means clustering (gray). Accuracy indicates the percentage of approximately 100 simulated treatments under each condition that were called accurately by each method. (
We next sought to compare the performance of the mp-value to other metrics designed to determine whether two multivariate samples are different. Traditional multivariate statistical tests such as MANOVA and Hotelling’s T2 test cannot be conducted when the number of readouts is greater than the total number of replicates, as in the simulated data set. Therefore, two other statistical tests (Student’s t-test and Wilcoxon rank-sum test) were performed for each single readout, with the resulting 100 p values being summarized to one p value by meta-analysis. The third method, k-means clustering, has been used previously in the analysis of multivariate biological data.22,23 All three of these tests performed similarly to the mp-value in correctly identifying negative controls, with the t-test performing worse than the others ( Fig. 2A ). The decrease in the performance of the t-test would be amplified at the screen level if most treatments in a screen mimic the vehicle control; in this case, the t-test would produce far more false-positives than the other three tests. In addition, the t-test cannot be calculated for treatments in which only a single replicate is available.
For the positive control treatments, all metrics improved when more readouts were simulated to differ between the treatment and vehicle and when the magnitude of the difference was larger ( Fig. 2 ). Relative to the other tests, the Wilcoxon rank-sum test was outperformed for nearly all positive controls. The k-means clustering also performed quite poorly on the positive controls when compared with the mp-value and t-test ( Fig. 2B – D ). The mp-value and the t-test performed best on the positive controls, with the mp-value outperforming the t-test in calling true-positives when few readouts differed between the treatment and vehicle replicates. Because it also outperformed the t-test in correctly calling negative controls, we believe the mp-value is most appropriate for biological screens, which we have found in our experience to have few truly differing readouts and many treatments that do not differ from vehicle controls. For example, in a screen in which 4 of 100 readouts differ in 5% of the treatments (with the remaining 95% of treatments resembling the negative control), extrapolating from the data shown in Figure 2 suggests that the mp-value would be predicted to call 89% to 92% of samples accurately, depending on the magnitude of the biological difference, whereas the t-test would call 78% to 80% correctly. Because of these results, we are confident in the accuracy and the appropriateness of the mp-value for differentiating between pairs of treatments in real biological screening data.
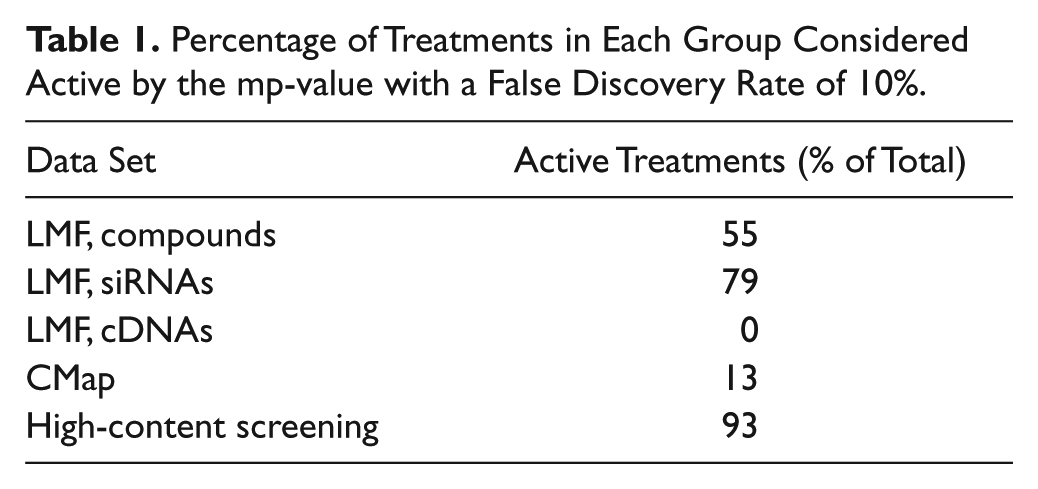
The mp-value calculation was first applied to the LMF data set, which consists of measurements of gene expression levels conducted using 100 probes representing 97 unique genes. Three classes of treatments were screened on this platform: compounds, siRNAs, and cDNAs. Of the 7052 compounds screened, 3893 (55%) had mp-values that were smaller than the statistical significance cutoff, suggesting that they produced a significantly different 100-probe profile than vehicle control and could be considered active in this screen ( Table 1 ). The percentages of active treatments differed for the genomic reagents. Similar numbers of siRNAs and cDNAs were screened on the LMF platform, but the percentages of active treatments were very different. Seventy-nine percent of siRNA treatments had statistically significant mp-values, whereas none of the cDNA treatments reached statistical significance using a false discovery rate threshold of 0.1. Many cDNA treatments tended toward significance; however, 25% of them had nominal mp-values less than 0.05.
Percentage of Treatments in Each Group Considered Active by the mp-value with a False Discovery Rate of 10%.
Several possibilities exist for the differential activity of the siRNAs and cDNAs. Importantly, cDNA selection was unbiased, whereas siRNA selection was limited to genes of interest to internal drug development programs. It makes sense that perturbing these genes might be more likely to produce signatures indicative of biological activity. Another explanation concerns off-target effects, which are known to be common with siRNAs.24,25 In a signature measuring 97 genes, a given siRNA may perturb several as a result of off-target activity. If these perturbations are consistent across replicates, the siRNA will likely have a statistically significant mp-value. In contrast, cDNA reagents lack a similar inherent mechanism for producing transcriptional off-target effects. Therefore, cDNA-induced perturbations of the gene signature may be comparatively rare. In addition, the negative cDNA control was moderately variable across replicates, and this increased noise likely also contributed to the lack of statistically significant findings.
To explore the biological validity of the mp-values, a detailed analysis was undertaken of a subset of the compounds screened in the LMF data set. In this subset, 256 compounds were run in four-point dose response at concentrations of 10 nM, 100 nM, 1000 nM, and 10 000 nM, and 131 compounds were run in eight-point dose response at concentrations of 5 nM, 14 nM, 41 nM, 123 nM, 370 nM, 1111 nM, 3333 nM, and 10 000 nM. Across all compounds, treatments had more statistically significant mp-values at higher concentrations ( Fig. 3A , B ). To determine whether this aggregate pattern held true for each compound, mp-values were plotted by dose for each compound and then subjected to linear regression. The results of this analysis, details of which can be found in the supplemental material, were highly statistically significant and indicated that mp-values tended to be more statistically significant at higher doses, as expected. These results strongly support the biological validity of the mp-value.
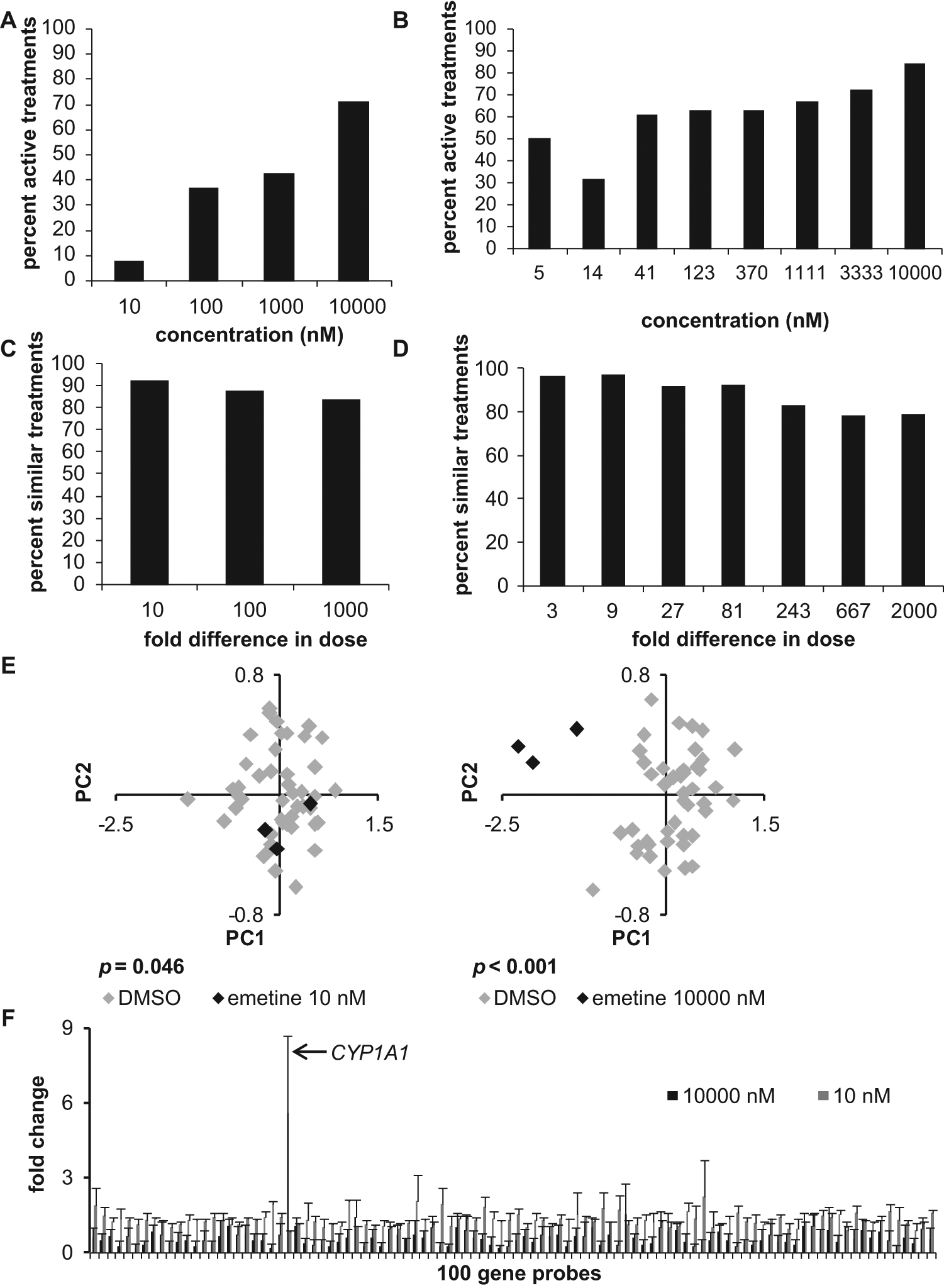
(
A second analysis of the LMF data set supported the ability of the mp-value to detect phenocopies and thereby answer the question of whether two treatments are similar. In large screens, one often wants to identify treatments that act similarly to a positive control, even if the potencies may differ. For example, readouts obtained for a given compound at the 10 nM dose would be expected to be more similar to readouts obtained at 100 nM (a 10-fold difference in concentration) than to readouts obtained at 10 µM (a 1000-fold difference). To test this in the LMF data set, replicates for each dose of a compound were compared with replicates of other doses for that same compound. As shown in Figure 3C and D , two treatments were more often indistinguishable by the mp-value when the dose difference was smaller. However, even when the difference in two doses of the same compound was large (i.e., 2000-fold), the treatments were still called similar approximately 79% of the time. These findings are encouraging and suggest that phenocopies of a treatment can successfully be identified using the mp-value, even in cases in which there are differences in potency.
The mp-value’s biological validity was also supported by examining which compound classes tended to produce significant mp-values. Because LMF is a transcriptional readout, it would make sense for compounds that affect transcriptional processes to have statistically significant mp-values. The biological process terms for 1731 Gene Ontology (GO) were linked to at least three compounds in the LMF data set by using internal compound-target and target-GO term annotation. Of these, 445 GO terms were overrepresented by Fisher’s exact test in the subset of compounds considered active, using a false discovery rate threshold of 0.1. If compounds that target transcriptional processes tend to be more active in the LMF data set, then transcription-related GO terms should be enriched within the 445 GO terms found to be associated with active compounds. This was indeed the case; 30 of the original 1731 GO biological process terms were related to transcription, and 20 of these were enriched in the GO term subset associated with active compounds (
The reliability of the mp-value was also tested on another gene expression–based data set: the Broad Connectivity Map (CMap). The CMap data set contains many more readouts than LMF—more than 22 000 compared with 100. However, the subset of CMap analyzed here shares many similarities with the LMF data set: The cell line is the same (MCF7), as is the time point (6 hr). Of the 1150 compounds screened in this subset, 835 were also screened in LMF. Although many experimental parameters differ between CMap and LMF, there is still high concordance between the significance of the mp-values for the 835 compounds present in both data sets, with a p value of 2.899 × 10−7 by Fisher’s exact test for the contingency table shown in Table 2 .
Agreement of the Significance of mp-values for Compounds Present in Both the LMF and CMap Data Sets.

Of the 1150 CMap compounds analyzed, 144 (13%) had statistically significant mp-values. This percentage is substantially lower than the 55% of compounds active in the LMF data set. When the percentages are calculated using only the 835 compounds that were screened in both this subset of CMap and in LMF, the percentages are slightly more similar; 15% are active in CMap, and 38% are active in LMF. A possible explanation for the difference is that the vehicle replicates for CMap may have exhibited higher variability than the vehicle replicates for LMF. In CMap, each treatment was usually run only once in a given batch, so batches were combined to have sufficient replicates to calculate mp-values. Normalization methods performed well in reducing batch effects (
The CMap data set also presented an opportunity to use the mp-value methodology to answer a different type of question: whether two noncontrol treatments are different from each other. In this data set, eight microarrays were run using trichostatin A obtained from Calbiochem (La Jolla, CA), and 51 microarrays were run using trichostatin A from Sigma (St. Louis, MO). Aside from being run in a number of different batches, all other experimental parameters were identical for these 59 arrays. A plot of the first two principal components for these treatments is shown in Figure 4 . The mp-value calculated for the comparison of these two groups was less than 0.001 and highly statistically significant. This strongly suggests a fundamental difference in the chemicals from two different suppliers.
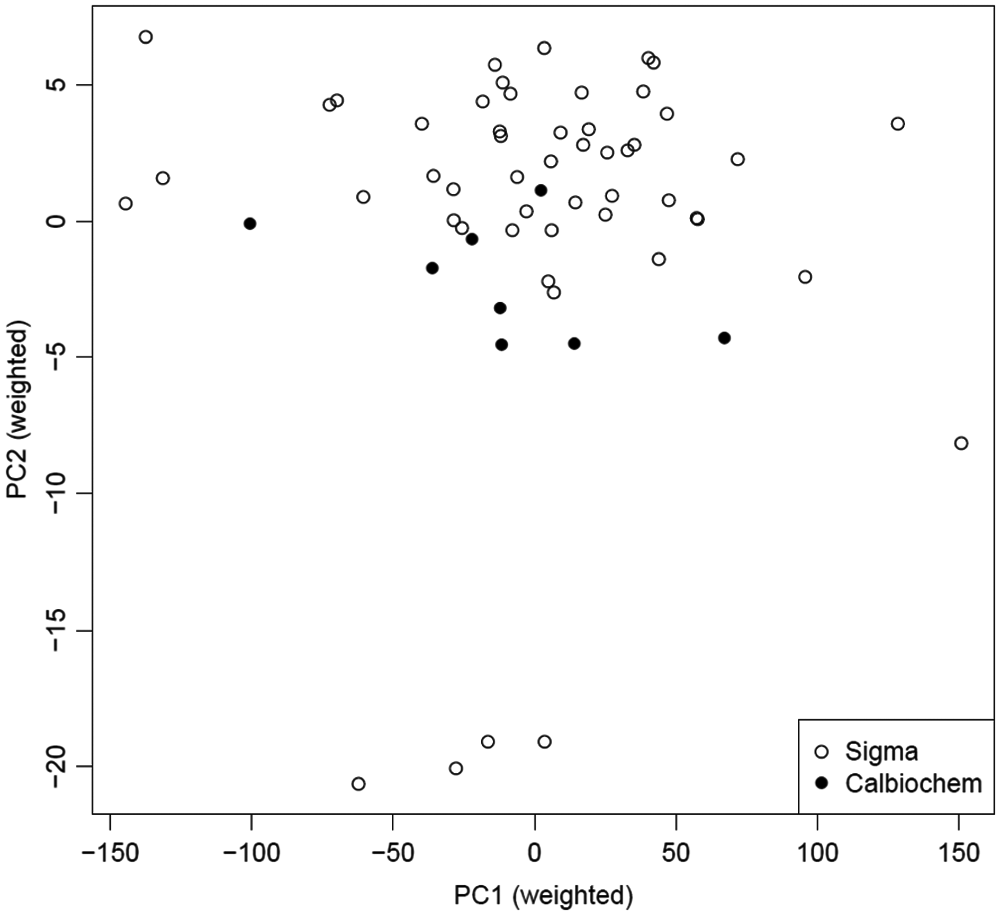
Plot of the first two principal components describing the subset of CMap data containing the replicates using trichostatin A supplied from Sigma and the replicates using the same chemical supplied from Calbiochem. The mp-value calculated using these data is statistically significant (mp-value <0.001).
The mp-value was next used to evaluate treatments in a third data set, an HCS of natural products. In this data set, 92.7% of treatments had statistically significant mp-values, a much higher percentage than was found for either of the gene expression–based data sets. A likely reason for this is that there was much more statistical power in this data set because of the high number of replicates, as hundreds of cells were imaged for each treatment and considered as individual replicates. With this sample size, treatment-vehicle differences that are small in magnitude can still be identified as statistically significant. Indeed, many of the treatments in this data set with significant mp-values had much smaller Mahalanobis distances than those seen in the LMF and CMap data sets (
The mp-value is intuitive, straightforward, and easy to calculate. To our knowledge, it is the first method that fully uses all readouts and replicates obtained in a screen and assigns a statistical significance value to each treatment based on an easy-to-visualize separation. Others have developed metrics that use the Mahalanobis distance, PCA, or similar methods for describing the similarity of multivariate profiles, but we believe this to be the first method to use these procedures in concert and to provide an estimate of statistical significance.27–31 Previously published methods also often require scientists to make an a priori selection of relevant readouts, samples for training set, or a cutoff for the final metric.27–31
The mp-value can be applied to data from many types of multidimensional screens; we have demonstrated its use here on two gene expression data sets and one high-content imaging data set. These data sets contained varying numbers of readouts (29, 100, and more than 22 000). The biological validity of the mp-value was demonstrated in several ways. As expected, it identified a higher percentage of active treatments among the highest doses versus the lowest doses for compounds run in dose response. In addition, it detected expected effects on transcription in a gene expression-based data set. It also had a high concordance rate when evaluating the significance of a compound in two different expression-based data sets.
As the results from the LMF, CMap, and HCS data sets have shown, there are several key factors that contribute to whether an mp-value will be statistically significant. The first and most intuitive of these is the magnitude of the difference between the two experimental groups; the larger the biological difference, the higher the likelihood that the mp-value will be statistically significant. The second factor is the variation in the replicates for each experimental group. As mentioned above, increased variation in replicates of a given treatment can cause an increase in the mp-value. Including replicates from multiple experimental batches can introduce such variation, so we have stressed that analyses should compare treatments run in the same batch. Finally, the third major factor is the number of replicates that are run. As shown in the HCS data set, when hundreds of replicates are available for each treatment, mp-values can be statistically significant even when the biological differences are small. The converse is also true; with insufficient replicates, mp-values for truly different treatments might not reach statistical significance. We recommend that experimental treatments within a batch be run in duplicate at minimum (and preferably in triplicate) and that controls be run in higher numbers (preferably at least 8 to 16 replicates per batch). Consideration of the first two factors mentioned above (magnitude of expected biological difference and expected assay variation) during assay planning will help inform the selection of the number of replicates to run for each treatment.
Despite the strengths of the mp-value, there are characteristics of the mp-value that may present challenges in certain situations. In some cases, such as in the HCS data set tested here, nearly all of the assayed treatments may have statistically significant mp-values. The mp-value itself is designed to merely say whether two treatments are different; its value should not be used to further prioritize this subset of significant treatments. However, as mentioned above, the Mahalanobis distance calculated during the mp-value calculation process can be used for a secondary prioritization, as it indicates the magnitude of the difference between the treatments. The provided R code outputs both the mp-value and the Mahalanobis distance so both metrics can be considered if desired. The permutation process introduces another characteristic of the mp-value that some may find inconvenient; it produces a limited number of discrete mp-values equal to the number of permutations run. To avoid this, a distribution can be fit to the Mahalanobis distances produced by the permutations, and the p value of the original Mahalanobis distance can be calculated according to the parameters of that distribution. We tested this approach in the HCS data set using gamma distributions fitted for each and found a high correlation between the original mp-values and those derived using the gamma distribution parameters (
One final inherent characteristic of the mp-value is that its distribution can vary widely from batch to batch. In one batch, vehicle replicates may be highly similar to each other, leading to more significant mp-values than would be seen in a batch in which vehicle replicates are not as consistent. The batch-to-batch variability of mp-values may be seen as an inconvenience, but it can also be used to indicate assay quality; if a batch has no statistically significant mp-values, it may be useful to investigate whether there was a consistency issue with the negative controls. When comparing the positive control(s) to the negative control(s) in a given batch, the mp-value can be considered to act similarly to the univariate Z′ factor; both provide a metric describing the difference between positive and negative controls. 32 In general, we find the mp-value to be a simple, appealing solution for evaluating treatments while dealing with the issues of difference, variability, and reproducibility that are inherent in multidimensional screening data.
Footnotes
Acknowledgements
We thank Fritz Roth for helpful discussions and mentorship. We would also like to acknowledge Yan Feng, Anne Kummel, and Xian Zhang for providing the HCS data set and accompanying annotation. In addition, we would like to thank Chris Wilson, Christian Parker, Xian Zhang, and John Tallarico for valuable feedback and discussions. Finally, we wish to thank those who have read and provided feedback on this article, including anonymous reviewers. Janna Hutz and Florian Nigsch are the recipients of NIBR Presidential Postdoctoral Fellowships.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.