
Abstract
The drug discovery landscape is littered with promising therapeutic targets that have been abandoned because of insufficient validation, historical screening failures, and inferior chemotypes. Molecular targets once labeled as “undruggable” or “intractable” are now being more carefully interrogated, and while they remain challenging, many target classes are appearing more approachable. Protein tyrosine phosphatases represent an excellent example of a category of molecular targets that have emerged as druggable, with several small molecules and antibodies recently becoming available for further development. In this review, we examine some of the diseases that are associated with protein tyrosine phosphatase dysfunction and use some prototype contemporary strategies to illustrate approaches that are being used to identify small molecules targeting this enzyme class.
Introduction: The Society of Drug Targets
Ever since the days of Paul Ehrlich, we have mechanistically viewed drugs as small keys fitting snuggly into much larger locks now labeled targets. It has been estimated that for the >1500 unique drugs approved by the U.S. Food and Drug Administration, there are at least 800 molecular targets. 1 We know that some drugs, such as the osmotic diuretics and the general anesthetics, lack well-defined targets, while others, such as metformin and valproate, have debatable molecular targets. For the remaining drugs, however, the endogenous macromolecules with which the drugs interact and mediate their desired therapeutic actions are almost always proteins, although some drugs target DNA and chelating agents trap metals. We loosely classify the protein drug targets into three major groups. The most common drug targets are enzymes and transporters that exhibit aberrant activation or elevated expression levels in human diseases. These targets demand selective inhibitors, generally blocking the catalytic activity of the enzyme or the cargo of the transporter. The protein tyrosine kinases represent one example from this group that has seen rapid growth in the number of clinically approved inhibitory drugs, especially in oncology. 2 Remarkably, however, almost 70% of the kinase family members remain unexplored as drug targets. 3 The largest drug-targeted family is the rhodopsin G-protein-coupled receptors (GPCRs), with 79 being bona fide therapeutic targets and another 46 being explored. 3 Nevertheless, 58% of the GPCR family also remain drugless and unstudied. This supports the notion that as drug discoverers and developers, we tend to gravitate to a limited number of molecular targets. 4
A second, less drug-friendly group of potential therapeutic targets are macromolecules, including proteins, which elaborate a loss of some biological or biochemical function. In this case, a small molecule, generally <1% of the mass of the target, must restore a lost function. For example, Duchenne muscular dystrophy is caused by genetic mutations in the protein dystrophin, which results in progressive muscle degeneration and weakness. Conceptually, it is quite challenging to design small molecules that would reinstate the complex function of dystrophin with a mass of 427 kDa. Nonetheless, activators of either normal or mutated forms of proteins and downstream events have been identified. 5 Another example is cystic fibrosis, which is an autosomal recessive disease caused by a mutation in the gene encoding the 170 kDa cystic fibrosis transmembrane conductance regulator (CFTR). There are ~1500 potentially causal mutations in CFTR, but >90% of U.S. patients have the F508del mutation, which creates a protein that does not fold normally and is not appropriately transported to the cell membrane. This results in its inappropriate degradation. There are two innovative drugs approved by the U.S. Food and Drug Administration for cystic fibrosis that target the pathological aspects of the F508del mutation: ivacaftor and lumacaftor. 6 Ivacaftor binds to the defective protein at the cell surface, opens up the chloride channel, and restores the proper flow of fluids and sodium. Lumacaftor facilitates the translocation of the defective CFTR protein to the cell surface. This example illustrates beautifully how understanding the molecular pathogenesis of a loss-of-function macromolecule can be translated into innovative therapeutics.
There are at least 25 well-documented tumor suppressor genes involved in the promotion and maintenance of human cancers, including PTEN, TP53, APC, VHL, BRCA1, PP2A, and Rb. Loss of the functional products of these genes is a major contributor to a large number of human malignancies, and replacement of the lost function with small molecules is a monumental therapeutic challenge. Remarkably, small molecules have been identified that restore the lost functionality for some tumor suppressors. For example, Foster et al. 7 identified a prototype compound that both enabled mutant TP53 to maintain a conformation that restored DNA binding and stabilized wild-type TP53 DNA binding. This tool compound permitted the accumulation of transcriptionally active protein, which slowed in vivo tumor growth. 7 The serine/threonine phosphatase PP2A regulates many cellular processes and suppresses tumor growth. 8 Recently, small-molecule activators of PP2A phosphatase have emerged that provide exciting and innovative avenues for the restoration of dysregulated PP2A. 9
A third group of validated disease-associated drug targets are those that have been labeled “undruggable” or, more mildly “intractable,” and thus are orphans in the field of drug discovery. Some high-profile disease-associated proteins have been subjected to intense investigation for inhibitors without success, so they have passively fallen into this category. As articulated in a previous review, 1 we believe the members of this group are more appropriately viewed as “undrugged,” because of emerging structural information, new reagents, automated methodologies, expanded chemical libraries, and innovative computational tools. Table 1 provides a limited list of molecular targets that have received the undruggable moniker but we now have tool or probe compounds for these targets, which might be employed as initial leads for drug development. One famous example is Ras, which has been known to be an oncogene for more than 30 years. 10 It is involved in at least one-third of all human cancers, yet there are no clinically approved drugs that directly target the oncogenic forms of Ras. This is due, at least in part, to the absence of any cavernous hydrophobic site suitable for drug docking on the surface of Ras. Consequently, alternative strategies have been designed to dismantle Ras hyperactivation in cancer, but it remains to be seen if they can be reduced to a clinically useful entity. 10
Examples of Historically Viewed Undruggable Targets.

Transcription factors, which form multiprotein complexes and coordinate gene expression, were thought to be undruggable because they employ energetically low, flexible, multicontact interactions between the transcription factor and DNA that spanned long distances. Conventional wisdom cautions that it would be improbable for a small molecule to efficiently disrupt such an interaction, 11 even though we know a change in a single amino acid or nucleotide can markedly diminish the binding and functionality of the complex. Thus, we probably should not be surprised that small molecules selectively disrupting transcription factors are now emerging.12,13 First-generation Myc transcriptional inhibitors were identified as disruptors of the Myc-Max heterodimeric complex using relatively small chemical libraries and either an in vitro fluorescence resonance energy transfer screen 14 or a yeast two-hybrid screen. 15 Although none of the initial hits are in clinical trials, these tool compounds and their refined analogs have been valuable reagents to investigate the biological roles of Myc. 12 Another example of an aberrant transcription factor that has been tamed by a small molecule is the fusion protein CBFβ-SMMHC, which is expressed after a chromosome inversion. CBFβ-SMMHC usurps the normal client protein for RUNX1, which deregulates RUNX1 transcription factor activity in hematopoiesis, and causes acute myeloid leukemia. A chemical library screen followed by lead optimization resulted in a bivalent small molecule, AI-10-49, which inhibits the binding of CBFβ-SMMHC to the RUNX1, restores RUNX1 transcriptional activity, and delays leukemia progression in mice. 13
It is widely recognized that most proteins interact with other proteins within cells in a nonenzymatic manner, and that these interactions regulate numerous signal transduction pathways. Abnormal protein–protein interactions are coupled to many human diseases.16,17 Blocking or disrupting these protein–protein interactions represents a major biochemical challenge, because the interface covers a large surface area, often lacks defined pockets, and comprises weak processive contacts.18–22 Advances in our knowledge about the precise nature of the interactions from x-ray cocrystals with ligands, the availability of more sophisticated chemical libraries, and the use of fragment-based nuclear magnetic resonance (NMR) molecule inhibitors of BCL-2 heterodimerization, such as navitoclax and venetoclax, have received U.S. Food and Drug Administration approval for use in the treatment of cancer.21,23–27 An innovative inhibitor of MCL1, S63845, was recently described with an impressive preclinical profile for both hematological and solid tumors. 28 There is little doubt that these prototypes will be emulated with many other disease-associated protein–protein interactions. 16
League of Tyrosine Phosphatases and Their Importance in Diseases
Many human diseases are the consequence of aberrant cell signaling. The most widely employed cellular signaling pathways are controlled by the addition or removal of covalent phosphate moieties. No molecular target class has seen greater clinical drug approval growth than the protein tyrosine kinases, with more than 28 being approved by the U.S. Food and Drug Administration. In contrast, the proteins that counterbalance the actions of tyrosine kinases, phosphatases, have generally been eschewed as drug targets. In particular, the protein tyrosine phosphatases (PTPs), which are ancient enzymes evolving from the protist Dictyostelium, represent a relatively large family comprising an estimated 125 distinct proteins in humans, which are divided into three major groups based on the amino acid catalytically involved in the removal of the phosphate from the substrate. The PTPs are further subgrouped by their protein sequences and functionality. It is noteworthy that despite their name, many of the PTP family members are endowed with the ability to remove a phosphate from serine, threonine, inositide, and carbohydrate, and mRNA. Assignment to the Cys-based PTPs, which is the largest group comprising three classes and six subclasses ( Fig. 1 ), requires the presence of the conserved canonical active site sequence, (CXXXXXR), which encompasses the catalytic cysteine and arginine bordering five variable amino acids ( Fig. 1 ).29–33 There is considerable evidence that Cys-based PTPs are significantly involved in many human diseases,30,34–38 as illustrated by the examples presented in Table 2 . Initially, however, PTPs were thought to be constitutive enzymes with little regulation, making them poor drug targets. 39 Multiple laboratories have dispelled this concept, showing a variety of PTP regulatory mechanisms, including transcriptional, translational, epigenetic, and posttranslational modifications; degradation; subcellular localization; and oxidative control. The notion that any substrate competitive inhibitor would need to emulate the charged nature of the phosphorylated substrate and thus have difficulty entering cells also repelled drug hunters. The lack of readily available potent and selective small-molecule inhibitors of PTPs, as well as a limited number of x-ray crystallography and NMR structures, further restricted efforts to develop tool compounds and therapeutic leads. A growing number of PTP structures have altered the landscape, providing several specific and potent small-molecule PTP inhibitors and making the PTPs now seem quite druggable. 40 The goal of this review is to focus on recent advances in identifying promising PTP inhibitors, amplifying the comments found in several reviews that previously discussed the challenges of targeting PTPs.1,31,32,37,40,41
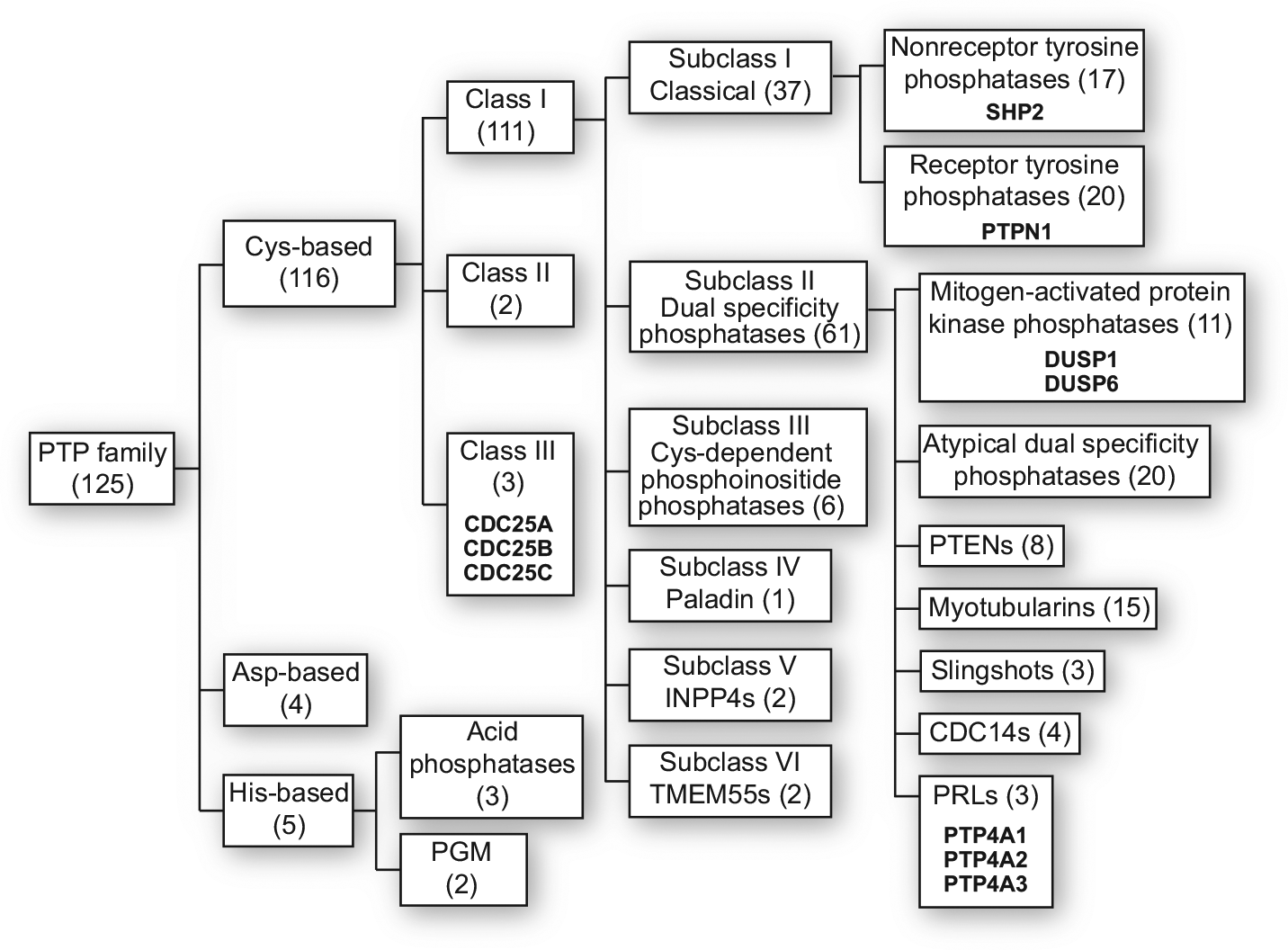
Human PTP classification. Members of the family are indicated in parentheses. PTPs mentioned in the text are highlighted in bold.
Disease-Associated PTP Family Members.
By far the most popular lead compound discovery approach, built on the successful identification of tyrosine kinase inhibitors, has been the use of automated high-throughput in vitro screens with purified recombinant enzyme and artificial substrates that employed either fluorescent or colorimetric end points. Artificial substrates, such as para-nitrophenyl phosphate, 3-O-methylfluorescein phosphate, fluorescein diphosphate, and 6,8-difluoro-4-methylumbelliferyl phosphate, have commonly been used, because they are inexpensive, stable in aqueous solutions, and easy to adapt to automated high-throughput platforms, and they do not require prior knowledge about the protein substrate or its ability to dephosphorylate phosphorylated peptides in vitro. 42 Unfortunately, these artificial substrate-based high-throughput screens can be disappointing as a means to identify useful phosphatase inhibitor leads, because substrate specificity for the PTPs resides in binding to sites that are not the catalytic sites and substrates like fluorescein diphosphate exhibit complex kinetics. 43 Furthermore, the conformation of a number of PTPs, such as DUSP1, DUSP6, and SHP2, is markedly altered in the presence of the protein substrate, so only employing an artificial substrate obscures any conformational-dependent inhibition.44–46
The frequent failure of high-throughput screens may be due in part to the composition of the available chemical libraries. This has been an endemic problem in screening in general 47 but is especially acute for PTPs. Unlike kinases, which have catalytic sites that exhibit considerable selectivity for the peptide substrate and help to form the basis of focused chemical libraries that are built on ATP-based pharmacophores rather than the peptide substrates, the active sites of many phosphatases are quite permissive, rather shallow, highly solvated, and polar. Kinase inhibitor libraries are often populated with ATP analogs with limited diversity. Consequently, screeners quickly gravitated to small-molecule libraries that have been maximized for diverse chemical structures. Unfortunately, these chemical screening libraries are often populated with promiscuous “bad actors,” or so-called pan-assay interference compounds (PAINS), that are active in a large number of assays due to either their reactivity, ability to generate reactive oxidative species, micelle creation, or detection interference.48,49 PTPs are especially susceptible to electrophiles and oxidation because of their catalytic cysteine, which has a low pKa and is readily attacked. Future screens might benefit by examining the available dark matter compounds, which fail in a large number of assays and thus are not promiscuous inhibitors. 50 As outlined below, however, interesting small-molecule PTP inhibitors are emerging from the use of traditional high-throughput screening methods,51,52 and these chemotypes could form the foundation for new chemical libraries with a higher success rate. Computational screening with large virtual chemical libraries, especially when coupled with high-throughput wet-bench laboratory screening, has also generated some interesting small-molecule inhibitors,53–62 which could nucleate future libraries. Alternative strategies for the identification of small-molecule PTP inhibitors have included assays that are formatted to use cells, whole organisms, or computational methods. Outlined below are some prototypic examples of using innovative approaches to identify high-value chemical probes targeting PTPs, which could form the basis for future therapies. These examples were selected because they represent distinct branches of the Cys-based PTP family ( Fig. 1 ) 30 and are among the most advanced, potent, and selective PTP inhibitors reported to date.
Examples of Emerging Tyrosine Phosphatase Inhibitors
SHP2
The Src homology 2 domain-containing (SHP2) phosphatase is a classic type I intracellular nonreceptor PTP encoded by the Ptpn11 gene, and thus is also known as PTPN11. Activating mutations in SHP2 have been observed in many types of human cancer, and experimentally, it has been shown to be involved in many tumor processes, including cell growth and metastasis, making SHP2 the original oncogenic phosphatase. 63 Mechanistically, SHP2 appears to regulate numerous intracellular oncogenic signaling pathways, such as RAS-ERK, PI3K-AKT, and JAK-STAT, at least in part by its ability to bind and dephosphorylate RAS. 64 This attractive drug target has been interrogated by traditional high-throughput and computational screens, yielding some interesting candidate inhibitors of the catalytic site, including the anticancer drug estramustine phosphate.65–68 Unfortunately, there does not appear to have been much progress clinically with any of these compounds, likely due to poor pharmacokinetic properties, insufficient potency, lack of specificity among other PTPs, and other off-target effects. Consequently, focus has been redirected toward allosteric inhibitors of SHP2. Chen et al. 45 developed a screening strategy that exploited the conformational changes induced in SHP2 when a substrate binds and locks SHP2 in an autoinhibited conformation. This enabled the authors to sample conformational space at equilibrium between the active or open and the inactive or locked forms. Armed with a 100,000-compound corporate library, they identified 900 candidate inhibitors that were counterscreened against a truncated version of SHP2 that contained only the catalytic domain. This enabled the identification of an inhibitor binding to a novel allosteric site and circumventing the limitations associated with conventional active site–directed inhibitors. Two additional assays were used to prioritize six compounds that functioned as potential allosteric inhibitors. One of the six molecules, SHP836, was optimized further using structural information obtained from x-ray cocrystals and iterative chemical synthesis of analogs.46,46 The x-ray structures confirmed allosteric binding and provided a rationale for the intrinsic specificity of the second-generation lead compound SHP099, which had an IC50 of 70 nM against SHP2. In addition, the compound had high aqueous solubility, was orally bioavailable, and engaged SHP2 as indicated by inhibition of ERK phosphorylation and cancer cell line growth. Moreover, oral administration of SHP099 was well tolerated and dramatically reduced the presence of patient tumor-derived acute myelocytic leukemia cells in an orthotopic mouse model and the growth of subcutaneously implanted human squamous cell carcinoma xenografts, albeit with somewhat high doses of 75 and 100 mg/kg, respectively. 45 These results provide evidence that allosteric inhibitors could be valuable tools to investigate further the biological roles of SHP2, and they form the foundation for the development of future therapies.
PTPN1
PTPN1 (PTP1B) has a central role in regulating growth factor, hormone, and cytokine signaling pathways. 69 PTPN1 has oncogenic properties as its overexpression leads to the development of spontaneous breast cancer and it is required for HER2-induced breast cancer.70,71 Because PTPN1 has also been associated with diabetes, obesity, and Rett syndrome,72–74 there have been major attempts by both industrial and academic groups to identify competitive small-molecule inhibitors of PTP1B. One potent competitive inhibitor, CPT157633, was found to ameliorate the dysfunctions in motor skills and behavior in a mouse model of Rett syndrome. 74 The authors used CPT157633 to demonstrate that PTP1B directly targets the tyrosine residues within the TRKB kinase activation loop and therefore is a critical negative regulator of brain-derived neurotrophic factor (BDNF) signaling. They propose that elevated levels of PTPN1 cause BDNF resistance and are an important contributing factor to the etiology of Rett syndrome. The importance of BDNF in other neuropsychiatric conditions, including autism, further reinforces the potential to address a broad range of challenging diseases by inhibiting PTPN1. The phosphonate moiety in CPT157633, which confers the competitive inhibitory properties, will, however, likely limit the oral bioavailability that would be desirable for use in the treatment of Rett syndrome and most neuropsychiatric conditions. Nevertheless, this study provides cogent proof of principle for the use of small-molecule PTP inhibitors.
As an alternative to the challenges with directly targeting the catalytic domain, investigators have uncovered allosteric PTPN1 inhibitors that have appealing pharmacological properties. Most notable has been the discovery of MSI-1438, which targets the disordered, noncatalytic, carboxy-terminus of PTPN1. 75 Binding of MSI-1438 locks PTPN1 in an inactive conformation and effectively blocks enzymatic activity. The compound inhibits HER2 signaling in vitro and in vivo. At reasonable doses, namely, 5 mg/kg, the compound blocks the growth of established breast tumors and prevents lung metastasis in a breast cancer model. 75 These studies not only fortify the hypothesis that PTP1N is a valuable therapeutic target but also illustrate an attractive molecular approach to PTP inhibition.
DUSPs
The 10 enzymatically active human mitogen-activated protein kinase phosphatases dephosphorylate critical regulatory tyrosine, serine, and threonine moieties on the nodal signaling kinases, ERK, p38, and JNK, and belong to the large Subclass II dual-specificity phosphatase family ( Fig. 1 ). There is considerable evidence that the dual-specificity phosphatases, DUSP1 (also known as MKP1) and DUSP6 (also known as MKP3), are operative in human diseases, including cancer, cognition, depression, pain, and neurological development. 31 This stimulated a search for small-molecule inhibitors of DUSPs using traditional high-throughput approaches. A homogeneous 384-well fluorescence intensity high-throughput screening assay was designed and implemented for DUSP1 and DUSP6 as part of the National Institutes of Health Molecular Libraries Screening Center Network.43,76 The assays were performed in a robust manner for the high-throughput platform with Z′ factors of >0.5, and with low intraplate, interpolate, and day-to-day variability (CV < 20%). 43 A diversity library of >65,000 compounds was screened at 10 µM for inhibition of DUSP1 and identified 46 compounds with IC50 values of <50 µM and four with IC50 values of <1.0 µM. 76 Similar hit rates were obtained using the same chemical library and recombinant DUSP6, although the resulting compounds were unique inhibitors in each group. Unfortunately, the active compounds suffered from reactivity, poor solubility, or lack of cellular activity. Attention was therefore directed to alternative assay formats that would ensure cellular activity. First, a definitive high-content, single-cell screening assay was developed that used intact mammalian cells and quantitatively measured nuclear phosphorylated ERK in HeLa cells after a transient transfection with plasmids containing constructs that encoded a c-Myc-tagged DUSP1 or DUSP6 ( Fig. 2 ).52,77 With the plasmid concentrations that were employed, approximately half of the cell population was positive for c-Myc expression 24 h after transfection, and thus ectopic DUSP expression. A brief 15 min cellular exposure to agonists, such as growth factors or 12-O-tetradecanoylphorbol-13-acetate (TPA), stimulated ERK phosphorylation, which motivated its translocation to the nucleus. Because DUSP1 and DUSP6 aggressively dephosphorylate ERK, ectopic expression of these phosphatases suppresses nuclear localization. Only in the presence of a chemical inhibitor of the overexpressed phosphatase will agonist-induced nuclear localization be restored ( Fig. 2 ). Thus, we screened 720 natural products with the single-cell, high-content assay using cells that were transiently transfected with c-Myc-tagged-DUSP1. 52 Non-DUSP1-expressing cells in the same population were used as control cells. A two-dimensional Kolmogorov–Smirnov test was used with a z score of >3 to identify six positive compounds active in restoring nuclear phosphorylated ERK after TPA treatment. Among the six compounds, the most effective was sanguinarine, which was inactive in a parallel assay with DUSP6-transfected cells. Sanguinarine increased the phosphorylation of ERK in human pancreatic cancer cells and inhibited DUSP1 in vitro but was a less potent inhibitor against DUSP6, CDC25B, PTP1B, and VHR phosphatases. 52 Although multiple cellular activities and mechanisms of action have been attributed to sanguinarine, these results illustrate the value of using a robust high-content, single-cell platform to discover new inhibitors of phosphatases.
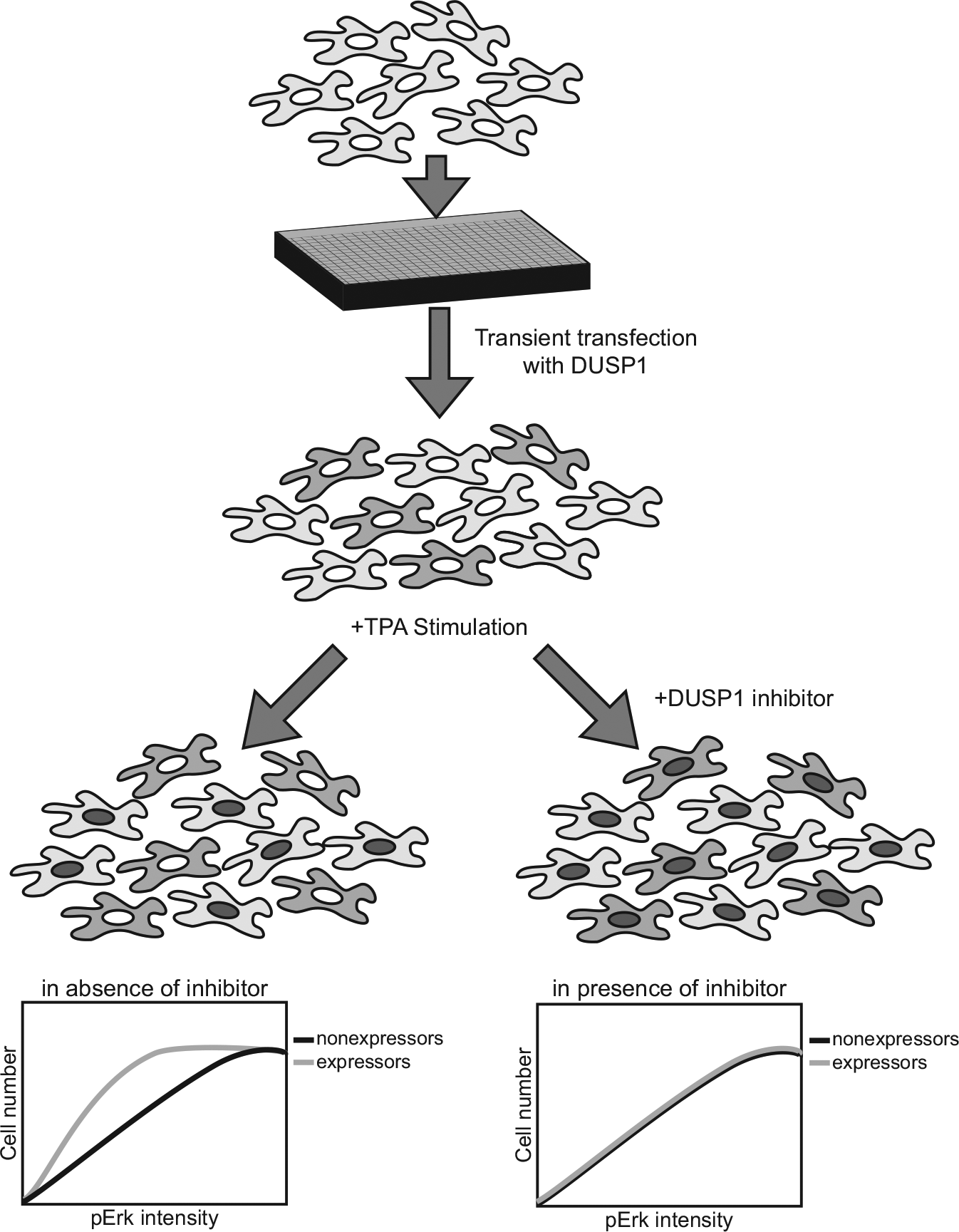
Schematic representation of a high-content assay for DUSP inhibition using HeLa cells. Adapted from Vogt et al. 52
Intact organisms can also be powerful platforms for discovering cell-active PTP inhibitors, albeit with lower throughput than population- or single-cell-based assays. The transparent zebrafish embryo is a vertebrate animal model that is ideally suited for high-content, small-molecule screening. The small size of a zebrafish embryo, its rapid development, and the ease of handling enable one to identify compounds that affect developmental processes and chemical modulators of signaling pathways in vivo. Fibroblast growth factor (FGF) regulates the development, proliferation, and cellular homeostasis by binding to tyrosine kinase receptors and activating intracellular signaling pathways, including those regulated by DUSPs. Reporters for FGF signaling have been generated, which allow for the live visualization of signaling activity during early development. 44 This whole organism model system was used to screen a small 5000-compound chemical library and 2-benzylidene-3-(cyclohexylamino)-2,3-dihydroindenone (BCI) was identified, which markedly enhanced FGF target gene expression in the zebrafish embryos. 44 An automated methodology has now been developed that enables much more rapid chemical screens with zebrafish. 78 Studies with mammalian cells confirmed BCI treatment increased phospho-ERK nuclear localization and blocked recombinant DUSP6 phosphatase activity in vitro. Of particular interest were the predictions from docking simulations that BCI was an allosteric inhibitor, which required the conformational changes stimulated by substrate binding. BCI had previously been screened in an in vitro assay and was not detected as an inhibitor. Indeed, incubation of recombinant DUSP6 with an artificial substrate in the absence of ERK produced no enzyme inhibition. BCI was used to demonstrate a temporal role for DUSP6 in restricting cardiac progenitors and controlling heart organ size at varying developmental stages. BCI has more recently been shown to overcome resistance to the tyrosine kinase inhibitor imatinib in a mouse model of acute lymphoblastic leukemia. 79 This is an example of how whole-cell or organism assays, which recreate the in situ environment, can expose valuable tool compounds. Indeed, second-generation BCI analogs have been reported to induce apoptosis in human breast cancer cells and sensitize them to lymphokine-activated killer cell activity. 80
PTP4A
Humans express three members of the phosphatase of regenerating liver (PRL) family: PRL-1, PRL-2, and PRL-3. Because PRL-3 phosphatase is not overtly expressed in the liver, the family is more accurately referred to as PTP4A phosphatases. They are a unique class of small, intracellular, prenylated phosphatases, which have high amino acid sequence homology: PTP4A1, PTP4A2, and PTP4A3 share ~80% amino acid sequence identity. There are differences in the expression of the isoforms, but we believe they share common biochemical functionality. Unlike most of the other PTPs, the PTP4A phosphatases activate the signal transduction mediators, ERK and AKT, which are central growth-promoting proteins in cancer. All three of the PTP4A family members are overexpressed in human tumors, stimulate cell migration and invasion, and increase in vitro replicative ability. 1 PTP4A overexpressing cells form tumors with high metastatic potential when injected into mice. 1 Consequently, the PTP4A family members are thought to be among the most oncogenic of all known phosphatases and highly valued therapeutic targets.
There have been several attempts to identify small-molecule PTP4A inhibitors. One of the earliest successes was a high-throughput screening assay by Daouti et al. 51 that used purified recombinant PTP4A3 and a bead-immobilized metal ion affinity-based fluorescence polarization assay with a phosphopeptide substrate. The authors screened a large Roche chemical library and identified a thienopyridone as a potent (IC50 = 128 nM) inhibitor of PTP4A3. 51 The compound also inhibited PTP4A1 and PTP4A2 with somewhat higher IC50 values. The thienopyridone did not inhibit 11 other phosphatases and had cellular activity. There is no information about its in vivo activity available. Nevertheless, follow-up studies have exposed new analogs, which are at least 50-fold more potent and hold promise as further leads for PTP4A inhibitors. 81 Computational approaches have also been applied to the PTP4A family despite the lack of a large number of x-ray crystal structures. 82 One provocative report by Hoeger et al. 54 used virtual screening approaches to identify novel small-molecule inhibitors of PTP4A3. They observed that ligand-based approaches afforded better results than docking-based techniques and reported that a 2-cyano-2-ene-ester, which was further optimized by a structure–activity relationship study, produced a low-micromolar PTP4A3 inhibitor with selectivity compared with three other PTPs in vitro. The compound reverted PTP4A3-induced cell migration in HEK293 cells. It also inhibited PTP4A1 and PTP4A2 in vitro.
Perhaps one of the most innovative strategies to identify inhibitors of PTP4A family members was conducted by Bai et al. 53 Because of the CAAX motif, the PTP4A family members are prenylated, and thus bind to membranes, which is essential for their function as oncogenes. PTP4A1 is trimeric in the crystalline state and in solution. 83 Previous studies suggest that trimer formation is essential for PTP4A1-mediated cell growth and migration. Moreover, the interface responsible for PTP4A1 homotrimerization is shared by the other PTP4A family members. This stimulated a computer-based virtual search for small molecules capable of disrupting PTP4A trimerization.53,83 Biochemical and structural analyses identified compound 43, which binds to the PTP4A1 trimer interface and blocks trimerization of PTP4A1, PTP4A2, and PTP4A3. 53 Compound 43 also specifically abrogated the PTP4A1-induced cell proliferation and migration through attenuation of both ERK1/2 and AKT activity. Additionally, compound 43 had anticancer activity in a xenograft mouse model of melanoma. The pioneering study provides strong pharmacological validation that trimerization is important for PTP4A function, and targeting it might be a viable approach for future therapeutic development.
Another interesting approach to inhibiting PTP4A3 has been the use of neutralizing antibodies. Although intracellular enzymes are generally not thought to be tractable targets for antibodies, there is growing evidence that they are accessible to antibody therapy or vaccination.84,85 In a series of publications,86–88 the Zeng laboratory has pioneered the use of exogenous mouse and humanized antibodies targeting PTP4A3 as a preclinical approach to treating melanoma, colorectal, ovarian, and gastric cancers. They have also successfully vaccinated mice with PTP4A3 and demonstrated a marked decrease in PTP4A3 replete tumor implantation in the immunized mice. 88 These studies could not only provide an exciting new alternative to targeting intracellular PTPs but also enhance the actions of small-molecule inhibitors.
CDC25
The Class III family of PTPs, CDC25A, CDC25B, and CDC25C, has been implicated in cancer and Alzheimer’s disease.37,89 All three members dephosphorylate tyrosine, serine, and threonine residues and have a critical role in the regulation of the cell cycle by activating cyclin-dependent kinase complexes. They are overexpressed in many human tumors. The CDC25 family members are activated in response to DNA damage, and deregulation of these checkpoint mechanisms can contribute to genetic instability and, ultimately, tumorigenesis. CDC25A and CDC25B have been reported to be overexpressed in the brains of patients with Alzheimer’s disease and are hypothesized to participate in aberrant neuronal cell cycle reentry, which leads to cell death and attendant neurodegeneration.90,91 Consequently, there has been a significant effort to identify small-molecule inhibitors of CDC25 phosphatases, particularly by using high-throughput screening methods.37,89,92,93 While many of the leads that were obtained from high-throughput screening suffered from the liabilities mentioned above for other PTP inhibitors, such as reactivity and generation of reactive oxygen species, some led to the more refined inhibitors with in vivo activity.94–96 While most of these compounds were irreversible inhibitors of CDC25, there has been some success in identifying reversible inhibitors that could have more promising pharmacological attributes. 97
Conclusion
Previously challenging drug target classes are now becoming exposed to small-molecule regulation due to the advances in assay systems, screening platforms, chemical library construction, structural information, and computational methodologies. The PTPs once carried the stigma of being undruggable, but recent developments of allosteric, orthosteric, and oligomerization inhibitors have revealed tool compounds that could form the basis of future therapeutics. 40 As more of these inhibitors become readily available, it is essential that each compound be fully credentialed and that we do not fall into the common pitfalls associated with some areas of preclinical therapeutic development, in which bold but unsubstantiated claims are made about compounds that lead others to expend time based on inappropriate conclusions. 98 In particular, there should be proof of target engagement using biophysical approaches, such as NMR, surface plasma resonance, and isothermal titration calorimetry. In addition, biochemical studies to determine the mode of inhibition, as well as site-directed mutagenesis, add confidence in the functionality of the PTP inhibitors. If we are mindful of these issues, we believe it is reasonable that the day will arrive when there will be as many phosphatase inhibitors approved by regulatory agencies for clinical use as there are protein kinase inhibitors.
Footnotes
Acknowledgements
The authors acknowledge financial support from the National Institutes of Health (R21 CA191944 and F31 CA196062), the Fiske Drug Discovery Fund, the Owens Foundation, the Cure Alzheimer’s Fund, and the Ivy Foundation.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors thank our past collaborators on the phosphatase projects: Andreas Vogt, Peter Wipf and Paul A. Johnston.