
Abstract
Production of novel soluble and membrane-localized protein targets for functional and affinity-based screening has often been limited by the inability of traditional protein-expression systems to generate recombinant proteins that have properties similar to those of their endogenous counterparts. Such targets have often been labeled as challenging. Although biological validation of these challenging targets for specific disease areas may be strong, discovery of small-molecule modulators can be greatly delayed or completely halted due to target-expression issues. In this article, the limitations of traditional protein-expression systems will be discussed along with new systems designed to overcome these challenges. Recent work in this field has focused on two major areas for both soluble and membrane targets: construct-design strategies to improve expression levels and new hosts that can carry out the posttranslational modifications necessary for proper target folding and function. Another area of active research has been on the reconstitution of solubilized membrane targets for both structural analysis and screening. Finally, the potential impact of these new systems on the output of small-molecule screening campaigns will be discussed.
Introduction
The discovery of small-molecule therapeutics during the past few decades has relied heavily on the production of recombinant proteins in sufficient quantity and quality for use in functional and affinity-based screening campaigns.1,2 Once small-molecule screening hits are selected by chemistry teams for further optimization, the focus shifts to activity in cellular assays, animal models, and eventually human testing, often with little regard to the properties of the target used to initially identify the molecules. Lack of efficacy in these downstream assays can occur for many reasons, including limited understanding of disease biology, 3 but any potentially significant differences between the endogenous target and the protein used for screening must also be considered, especially because the properties of the same protein expressed in different systems may not be similar.4,5 With the rapid proliferation of targets resulting from an explosion of genomic data combined with recent advances in affinity screening technologies that access significantly larger regions of chemical space than traditional screening decks,6,7 it is critical at the outset of any new screening campaign to consider expression technologies designed to produce proteins that structurally and functionally resemble endogenous targets. Such systems should also be more suitable for production of what have unfortunately been labeled as “challenging” targets, such as those that require complex posttranslational modifications or the presence of cell-type-specific protein partners to fold into their native conformation and multispanning membrane targets. 8 Production of these targets has been defined as challenging based only on limitations of the expression system chosen. Current estimates of the total number of human gene products that have been successfully targeted with small-molecule drugs range from 1% to 2%.9–11 Putting aside the debate of how much of the human genome is actually “druggable,” because this may dramatically change as new chemical space is explored,12,13 one can conclude that a significant number of disease-relevant proteins have yet to be successfully targeted using a small-molecule approach. 14 To date, crystal structures have been obtained for only approximately 15–20% of human proteins, but only slightly more than 2000 targets, largely protein kinases and other soluble cytoplasmic targets, have been co-crystallized with druglike molecules, 15 and thus there is a significant need for developing new expression and purification systems to drive both structural biology and production of novel screening targets. In this article, recent progress in expression-construct design, expression hosts, and reconstitution of membrane proteins will be discussed with an emphasis on how these new systems afford advantages in maintaining a close match between recombinant and endogenous targets and thus open up new challenging target classes for small-molecule screening efforts.
Expression-Construct Design
Many small-molecule drug-discovery projects begin by designing expression constructs of the screening target. Construct design is usually guided by the goals of the small-molecule screen. If the goals include identifying molecules that bind to novel sites on the protein, full-length constructs will be preferred, and the screen should be set up to detect all such binders. For example, the phosphoinositide-dependent protein kinase 1 (PDK1) contains three known small-molecule binding sites, the adenosine triphosphate (ATP) pocket, the phospho-transfer substrate pocket, and a site for binding to hydrophobic sequences in the C-terminus of kinase substrates known as the PDK1 interacting fragment (PIF) pocket. 16 Using the full-length kinase expressed in the baculovirus system and a substrate designed to be sensitive to binders at all three small-molecule binding sites, two new series of selective, non-ATP-competitive compounds were identified that may bind to the PIF pocket. 17 These novel binders would not have been discovered if only the PDK1 kinase domain was used or if the assay had been designed with a substrate that did not interact with all three sites.
If the goal of the screen is to target a particular binding site, then expression efforts should focus on producing the subdomain of the protein containing that site, as long as it is stable and functional in the absence of the remaining sequence. Such approaches have been used with a wide variety of targets, especially in cases when expression of the full-length protein has been challenging, and have recently been successfully used to identify small-molecule inhibitors of protein–protein 25 interactions. Many proteins involved in therapeutically relevant protein–protein interactions contain multiple domains, interact with multiple proteins in the cell, and can also form large multimers, all of which make expression and purification challenging. An example of such a target is the Cullin3 E3 Ligase, Keap1. Keap1 binds to Nrf2, a key regulator of cytoprotective genes, and targets Nrf2 for degradation by the proteasome. 18 Small-molecule inhibition of the Keap1–Nrf2 interaction may be beneficial in both oncology 19 and inflammatory disease. 20 Overexpression of full-length Keap1 in mammalian cells, however, results in the production of dimers and higher order multimers of the protein, 21 which bodes poorly for expression of this construct in nonmammalian systems. A domain based expression strategy, using Escherichia coli expression of the Keap1 Kelch domain only, produced high-quality monomeric protein. 22 The Kelch domain alone binds to Nrf2, and a recent screen conducted using a fluorescence polarization assay with the Kelch domain and a fluorescent Nrf2 peptide has led to the discovery of novel small-molecule inhibitors of this interaction that prevent Nrf2 degradation. 23
One of the most successful paradigms for expressing, purifying, and crystallizing proteins has been established by the Structural Genomics Consortium (SGC). To rapidly identify constructs of human genes that could be successfully expressed in E. coli and crystallized, they prepared full-length constructs and a series of truncated fragments of each gene. 24 They relied on the use of multiple bioinformatic approaches, such as PFAM-based 25 domain prediction and PSIPRED 26 secondary structure prediction, to define structured domains, but the primary approach was to use alignments with protein domains that had previously been crystallized. On average, a total of 9–15 constructs were designed for each domain by varying the boundaries of the bioinformatically defined domains by 2–10 amino acids. Successful expression was defined as the production of soluble protein in E. coli at expression levels greater than 2 mg protein/L of culture and up to as high as 50 mg protein/L. Interestingly, they found that only 11% (31 out of 282) of the constructs they tested could be expressed and purified as full-length proteins, some (6%) required the removal of nine or fewer amino acids, whereas the vast majority (80%) could only be successfully expressed with significantly larger truncations. 24 The obvious conclusion is that all expression projects should include truncated versions of the target as well as the full-length version to improve the likelihood of successful expression in E. coli. Other expression systems, however, should also be considered (discussed below), because another report on large-scale expression testing in E. coli has shown that the success rate of purifying soluble targets is only approximately 30% for both full-length native sequences and truncated synthetic sequences. 27
Furthermore, the SGC also found that the number of disordered residues at the ends of successfully expressed and crystallized constructs was on average very low, and for about 50% of the constructs all of the amino acids were in ordered structures. 24 The predicted number of disordered residues based on both PSIPRED- and PFAM-defined domain boundaries was much higher for both N- and C-terminal ends. The observation strongly suggests that basing new constructs on alignments with previously crystallized homologous domains should be much more successful than using secondary structure prediction. Obviously, this can be helpful only if related domains have already been successfully expressed and crystallized, and the disordered regions outside of those domains have been defined. If no prior related crystal structures exist, then beginning with the PFAM and PSIPRED domain definitions is a good start, but other constructs with varying N- and C-terminal extensions should be designed in parallel.
Gene Synthesis and Codon Optimization
Once constructs have been selected for expression, the appropriate clones must be generated. During the past few years, many labs have taken advantage of custom gene synthesis to generate expression constructs rather than lengthy subcloning procedures from cDNA libraries or from commercially available vector collections. 28 Producing constructs using gene synthesis provides a number of advantages over traditional cloning. Along with being less labor intensive, gene synthesis eliminates concerns over accumulating mutations that can occur during PCR-based amplification steps involved in the subcloning process. Gene synthesis also allows complete control over the selection of restriction sites used for subcloning the construct into the required expression vectors and the incorporation of affinity tags directly into the construct sequence at any location.
Another option that custom gene synthesis provides is the choice to optimize sequences based on the codon preferences of the expression host. Codon usage tables have been generated for many host organisms, including E. coli, based on various criteria, one of which is the presence or absence of certain codons in highly expressed proteins. 29 These tables are then used to alter the codon sequences of heterologous genes that are expressed in the host organism with the goal of maximizing expression levels. Simply replacing rare codons with more frequently represented codons does not, however, always increase expression levels. Analysis of more than 250 codon-optimized synthetic genes showed that for greater than 20% of the constructs, optimization had no effect on expression yields. 30 Other factors such as clustering of rare codons and the position of rare codons at the beginning of an open reading frame may be part of the reason why such codons have more deleterious effects than others on protein expression. 31 Preferences for an order of certain codons within constructs, observed as a bias toward one particular tRNA after it has been used for the first time in a sequence, may also be important for optimal expression. 32
In an effort to more fully understand the effects of rare codons in E. coli expression, a recent study assembled more than 14,000 synthetic sequences and tested the effects of rare codons in the N-terminus of expression constructs. 33 Surprisingly, the inclusion of rare codons in the N-terminus of a GFP (green fluorescent protein) construct substantially increased the expression of most constructs, in some cases as much as 100-fold. These results correlate well with the preference for rare codons in the N-terminus of highly expressed endogenous E. coli genes. The authors conclude that one significant reason for the rare codon preference in endogenous genes is that rare codons, which on the whole have lower GC content than more frequently used codons, produced more relaxed 5′ RNA structures that are translated faster by the ribosome. 33 Other studies have also clearly demonstrated the undesirable effects of 5′ mRNA secondary structures on protein production,31,32 but this is the first to do so by using such a massive array of synthetic sequences. A similar relationship between mRNA structure and codon bias in highly expressed yeast genes has led to the conclusion that the tRNA pool has adapted to the genomic codon profile rather than the other way around. 34 Further work needs to be done to determine the effects of such codons in other parts of constructs as well as to more fully understand the reasons for the N-terminus rare codon effect.
Finally, one must also consider recent reports on the effects of synonymous mutations before deciding to alter the native codons of a target. Because the presence and positioning of rare codons can slow down the rate of translation, 35 and because translation rate can have a significant effect on protein folding in E. coli, 36 replacing rare codons, especially at transition points in secondary structure, 37 may result in protein misfolding. Translation in E. coli is also slowed by reducing induction temperature to lower than the normal growth temperature of 37°C, and thus temperature reduction may give proteins more time to fold into their native structures. Synonymous mutations have also been shown to have effects on the specific activity of an enzyme, ADAMTS13, 38 and on the behavior of circadian clock proteins,39,40 and can even alter substrate specificity and small-molecule interactions with a membrane protein. 41 The potential for synonymous mutations to alter small-molecule binding is extremely relevant to both affinity- and activity-based screening, and it will be of interest to see how many other target classes are sensitive to codon alterations. Thus, although codon optimization is beneficial for specific targets, these benefits must be weighed against the small, but still possible, chance that synonymous mutations may cause the behavior of the construct to differ from wild type, and the more likely result that the “optimized” construct may not improve expression levels over the wild-type construct.
Fusion Tag Selection
Once the genetic material for the target protein is selected, the next major choice in expression construct design is the selection of a fusion tag. Fusion tags come in many different varieties and have been designed for a wide array of purposes.42,43 Two major purposes are to improve the solubility of recombinant proteins in E. coli and to provide a convenient handle for affinity purification.
The first class of tags includes TrxA, 44 based on the very soluble E. coli protein thioredoxin A; SUMO, 45 a small-ubiquitin-related modifier from yeast; and NusA, 46 a very hydrophilic E. coli protein that also promotes pauses in the coupled transcription and translation machinery in E. coli to allow more time for protein folding. 47 Because these tags are based on very soluble proteins, they are able to confer solubility onto less soluble target proteins. Recently, larger screens of abundant endogenous E. coli proteins have turned up more potential fusion tags for insoluble targets. In one such screen, 88 highly expressed E. coli proteins were fused to three challenging model targets: human β-defensin 2, human epidermal growth factor, and human erythropoietin. 48 Twelve of the 88 were found to successfully increase soluble expression and were then retested using a panel of cytokines. Certain tags were able to increase soluble expression up to 29-fold.
Other fusion tags, such as TrpE, 49 ketosteroid isomerase, 50 and PagP, 51 are extremely insoluble and decrease the solubility of the fused recombinant protein, resulting in the deposition of the fusion product into E. coli inclusion bodies. These approaches have been used to produce peptides that are toxic to E. coli. 52 Targeting these peptides to inclusion bodies prevents the peptides from killing their expression host and simplifies purification because they can be readily extracted from the insoluble portion of the E. coli lysate. This approach may also be used for other very toxic proteins or for proteins that are subject to degradation in E. coli. Typically, these tags are removed prior to working with the target protein by using very harsh conditions such as cyanogen bromide in acid, but recent work using metal–ion-catalyzed peptide bond cleavage 53 may provide a gentler route to the desired product. Interestingly, this approach has also been used for the production of a very challenging target, the N-terminal sequence of human cardiac troponin I, an intrinsically disordered protein, as a PagP fusion construct. 51 Ketosteroid isomerase fusions have also been used to produce fragments of G-protein coupled receptors (GPCR) transmembrane domains 54 that could be very interesting targets for affinity-based screening approaches.
The second major class of fusion tags is referred to as affinity tags because they are added to proteins so that they can be purified by using the affinity of the tag for a specific matrix ( Table 1 ). Affinity tags are also often used in protein–protein interaction screens as handles for the attachment of reagents to detect the interaction between two differentially tagged constructs using homogeneous time-resolved fluorescence-based assays. 55 Affinity tags are also critical for affinity-based screening methodologies that rely on capturing protein–small-molecule complexes to identify hits. 7
Protein Affinity Tags. Abbreviations: EC, Escherichia coli; I, Insect Cells; M, Mammalian Cells. NA, Not Available.
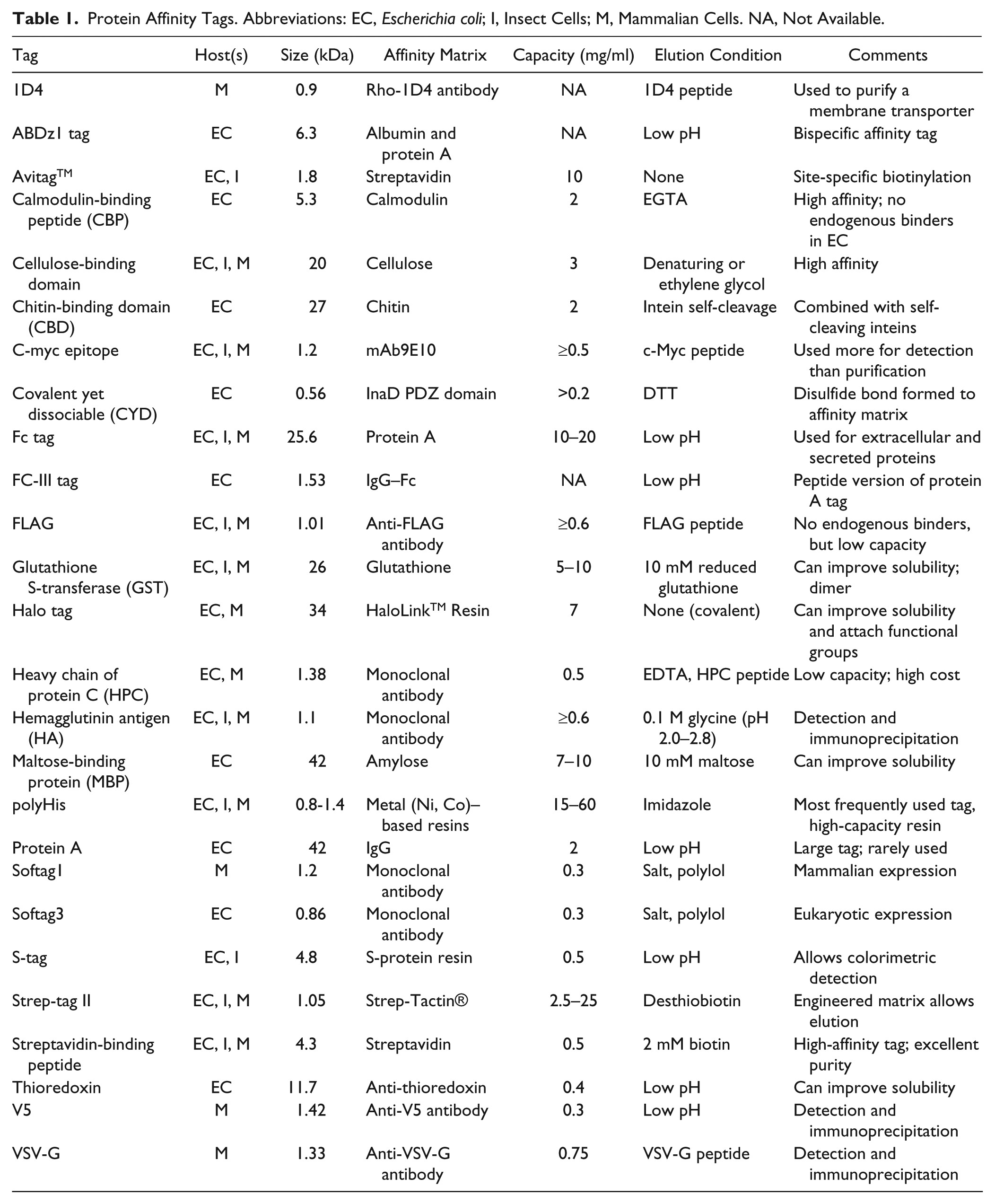
One of the most commonly used affinity tags on constructs today is the polyHis tag. Designed more than 25 years ago, the polyHis tag binds to metal-containing matrices and can be eluted with high concentrations of imidazole. 56 The polyHis tag, which can vary in length from 6 to 12 histidine residues, can be placed in almost any position in the protein sequence and, in most cases, will still be able to serve as an affinity handle for purification. Tag placement can, however, dramatically alter both the function of the tag and the behavior of the protein if the tag is required for the screening assay. 42 Just as multiple constructs may need to be tested to optimize protein expression, tagging constructs at both N- and C-terminal ends is also warranted, especially if the small-molecule site is proximal to one of the termini. It also may be necessary to test various linker peptide lengths between the tag and the beginning or end of the target gene. 57 Testing tags at both termini may also be important if the construct is going to be used in protein–protein interaction screens, and there is concern that the presence of the tag may sterically interfere with the interaction. PolyHis tags have been successfully used on a wide variety of targets, but they are not recommended for use with metalloenzymes because the polyHis capture matrix may extract metals from the active site and reduce specific activity. 42
Other popular tags include GST, MBP, FLAG, and Strep tags. GST refers to the 26 kDa glutathione S-transferase enzyme from Schistosoma japonicum that binds to immobilized glutathione and can be eluted by excess glutathione. 58 Maltose-binding protein, or MBP, is a hydrophilic 42 kDa E. coli protein that binds to immobilized amylose and can be eluted with maltose. 59 GST and MBP tags can often increase the solubility of a target, but because GST is an obligate dimer, 43 it may present the protein to the small-molecule screening deck in a nonnative conformation. The FLAG tag is an 8 amino acid hydrophilic tag that binds to several antibodies and can be eluted using excess FLAG peptide. 60 A side-by-side comparison of the FLAG tag to other proteins tags showed that it produced the highest level of purity from an E. coli lysate in a single-step purification. 61 Strep tags are based on peptides that have been selected from large libraries based on their ability to bind to streptavidin or streptavidin derivatives. 62 The Strep-II tag is a short 8 amino acid sequence that binds to streptactin, an engineered version of streptavidin, and is eluted with Desthiobiotin. 63 The longer streptavidin-binding peptide, or SBP tag, is a 38 amino acid tag that binds to streptavidin and can be eluted with biotin. 64 One disadvantage of using the SBP tag has been that the matrix was not reusable after the biotin treatment, because biotin binds essentially irreversibly to streptavidin. The recent discovery of a new streptavidin mutant that still maintains high-affinity binding to the SBP tag, but has significantly lower affinity for biotin so that the streptavidin matrix can be reused, may result in more frequent use of this novel tag on other proteins. 65 All of the tags discussed above can be engineered to be cleaved by site-specific proteases to remove the tags once purification is complete. 66 Tag removal is often desired for structural studies to prevent any tag-dependent artifacts, but it may also be important for screening campaigns that make use of reagents that bind to specific tags on targets.
Expression Hosts for Soluble Targets
The final major decision in the expression of a screening target is the choice of expression host. Due largely to its ease of use, potential for high protein yields, and commercial availability, E. coli, in particular the BL21 strain housing the lambda DE3 phage expressing T7 polymerase, is the most widely used expression host for both structural analysis and screening of protein targets. 8 Of the 85,577 structures deposited in the crystal structure database (http://www.pdb.org) that have annotations describing the expression host used, 75,592 or 88% were produced in E. coli. Only 3% of the crystallized proteins were produced in insect cells using the baculovirus system, and a mere 1.4% were purified directly from human tissue or produced using a human cell expression system. Analysis of protein sources used for co-crystal structures with druglike molecules also shows that the targets used to identify these molecules are also largely produced in E. coli, with a growing representation of targets produced in baculovirus. 15 Thus, to date, most proteins have been visualized and screened as represented by the E. coli expression system.
The caveats of E. coli expression for human targets have been described in many publications. Many target proteins, some have estimated as high as 77%, 67 are insoluble when expressed in E. coli, thus creating the need for the solubility-promoting tags described above. A specific example is interleukin-6 (IL-6), a secreted human protein that contains multiple disulfide bonds and naturally forms higher order multimers. 68 When IL-6 is expressed in E. coli, it is deposited in inclusion bodies, the destination of aggregated and often misfolded proteins, and purification of IL-6 from these inclusion bodies results in inactive protein. 69 Efforts to increase the solubility of IL-6 by fusing it to GST, MBP, and NusA only led to minor improvements in solubility and significantly increased the labor required to purify the final product. Targeting IL-6 to the periplasmic space did produce soluble protein, but the yields were dramatically reduced. Only co-expression of multiple cytosolic chaperones (DnaK, DnaJ, GrpE, GroES, and GroEL) combined with expression at room temperature and lower concentrations of the inducer, IPTG (isopropyl beta-D-thiogalactoside), resulted in reasonable levels of soluble cytosolic expression and bioactivity comparable to native IL-6. 69 Even though there have been some successful attempts to produce disulfide-bonded proteins in E. coli using various strategies 70 other than the multiple chaperone approach required for IL-6, some of these attempts have unexpectedly resulted in the production of periplasmic inclusion bodies 71 and expression of proteins with nonnative disulfide bonds, 72 as was most likely the case for IL-6. 69 Thus, the limitations of E. coli for the expression of human proteins can be overcome but only by testing multiple expression conditions, including localization and co-expression of chaperones.
Many limitations of the E. coli system for producing human protein targets can be overcome by switching to the baculovirus system, a eukaryotic system based on lepidopteran cell lines from organisms such as the fall armyworm, Spodoptera frugiperda. 73 The baculovirus system is the most widely used eukaryotic expression system for both screening and structural studies. 74 S. frugiperda cells contain posttranslational modification systems and chaperones similar to those observed in mammalian cells, and they have been used to produce soluble and functional forms of many recombinant human targets, including secreted proteins, such as interleukins, and transmembrane proteins. 75 The production of proteins using the baculovirus system is a two-step process. First, the target gene is packaged into the baculovirus particle by co-transfecting insect cells with the plasmids required for target and virus production. Several rounds of virus production are often required to attain the titer required for high protein expression. The virus is then added to the insect cells in suspension cultures to produce the cell mass needed for high protein yields. Although baculovirus protein production is more labor intensive than the E. coli system, the resulting yields of soluble and functional targets are often well worth the extra effort. It is not, however, the ideal system for all targets, because some posttranslational modifications, such as N-linked glycosylation, are slightly different from those of mammalian cells, 73 and these observations have driven recent efforts to reengineer the insect cells to produce fully mammalian sugar chains. 76
An alternate expression system for human targets is the methylotrophic yeast, Pichia pastoris. A recent publication comparing the expression of human adiponectin in both E. coli and P. pastoris systems demonstrated that the P. pastoris system produced more soluble protein per liter of cells than E. coli. 77 Only the P. pastoris protein contained posttranslational modifications and was able to demonstrate a more pronounced effect on mouse blood glucose levels than the E. coli material. 77 Another recent publication has demonstrated the superiority of the P. pastoris system over E. coli for the production of PDGF-AA. 78 Production of the N-terminus of urokinase plasminogen activator in P. pastoris also yielded biological active material that contained posttranslational modifications not seen in E. coli–produced protein. 79 Overall, P. pastoris is superior to E. coli for the production of bioactive proteins that require posttranslational modifications, particularly glycosylation. Strain engineering with P. pastoris has identified combinations of mutations and overexpression of metabolic enzymes that can result in significantly higher production of recombinant proteins than wild-type strains. 80 The P. pastoris expression system has been recently used to screen for inhibitors of human matrix metalloproteases (MMPs) that are very difficult to express as active enzymes in prokaryotic systems due to their mechanism of activation. 81 MMP2 and MMP9 were both expressed on the cell surface of P. pastoris by fusing the target constructs to a cell wall anchor sequence, and the intact cells were used as the enzyme source for a screen with a fluorescent substrate that resulted in the discovery of potent inhibitors. 81
Interestingly, descriptions of expression of human proteins in mammalian systems, and human cell–based platforms, are somewhat rare in the screening literature and in the online database of crystallized proteins. Even though such systems have been commercially available for some time, their use has often been limited to the production of antibodies. 82 Because of the limitations of E. coli expression for many full-length human proteins and because eukaryote-specific posttranslational modifications are often required for proper folding, new systems that readily allow side-by-side expression of protein targets in several eukaryotic systems have been developed. One example is the pFlp-Bac-to-Mam system, a single-vector system designed for expression in three host systems: baculovirus, transient expression in suspension culture–adapted HEK293 human cells, and stable expression in Chinese hamster ovary (CHO) cells. 83 The vector contains an Epstein–Barr virus oriP for the transient expression of the target in mammalian cell lines such as HEK293E, sites for Tn7 transposition into a bacmid for insect cell expression, and Flp–recombinase sites so that the insert can be rapidly integrated into certain CHO cell lines for stable expression. The authors compared the expression of three different proteins using this system. They found that the intracellular fluorescent protein, mCherry, was produced at very high levels of more than 50 mg/L in both the HEK293 and insect cells, with about fivefold lower expression in the CHO line. 83 The extracellular domain of the TL2 receptor was produced to nearly 1 mg/L in the CHO line, and scFc–hIgGFc was produced at 90 mg/L in HEK293 cells but only at 2 mg/L in the insect cells. 83 The advantage of this system was that all three expression systems could be explored in parallel in less than 12 weeks without the need for further subcloning or constructs design. The varying results with three different proteins clearly show the utility of such a system for quickly selecting the optimum host for expression of a screening target.
One of the challenges of expressing proteins in mammalian cells, such as HEK293 cells and their derivatives, is that they grow much more slowly than E. coli, and the assessment of multiple constructs using large-scale cultures can be quite laborious, especially if stable cell lines are generated for expression. To overcome these challenges, higher throughput methods for optimizing expression in mammalian cells have recently been developed. One system uses PCR to rapidly generate tagged expression constructs that are then tested for expression using small (2 ml) culture volumes in 24-well blocks, a dot blot apparatus, and a fluorescent antibody to detect the tagged protein after it is blotted on a membrane. 84 The expression of proteins in small culture volumes was found to be representative of expression in larger culture volumes and greatly accelerated the process of identifying highly expressed clones. A fully automated system for transient transfection of mammalian cells to assess protein production in a high-throughput manner has also been developed. 85
One example of a target for which the HEK293-based expression system has been successfully used for both structural and screening purposes is the secreted glycoprotein autotaxin. Autotaxin is a target for both new oncology and antifibrotic drugs, 86 but progress in building detailed structure–activity relationships had been limited due to the inability to produce the protein in enough quantity for structural studies. 87 Expression of autotaxin in HEK293 cells and purification of the secreted protein from the media have since been used to determine the crystal structure of the murine enzyme 88 and to successfully identify potent inhibitors by high-throughput screening. 89
Novel host organisms for the expression of human proteins have also begun to appear in the literature. Leishmania tarentolae, a nonpathogenic protozoan parasite of the white-spotted wall gecko, has been used to successfully express both soluble cytosolic proteins, 90 and secreted proteins, such as erythropoietin, that require mammalian-type high-mannose N-glycosylation. 91 Interestingly, this system makes use of the high transcription rate of RNA polymerase I by using vectors that integrate the recombinant expression cassette into a rRNA locus in the genome. 90 Insertion of 3′UTRs from highly expressed L. tarentolae genes can also greatly increase expression level. L. tarentolae can also perform other posttranslational modifications such as the incorporation of disulfide bonds and the processing of signal sequences, and it can handle more complex protein-folding challenges such as proteins with epidermal growth factor–like domains. 92 Human SOD1, without any additional fusion tag, was recently crystallized from protein expressed in L. tarentolae, demonstrating the ability of this system to produce large amounts (30 mg/L) of high-quality protein. 93 Other complex proteins that have been challenging to express in E. coli, such as the hepatitis E virus capsid protein, 94 have been successfully expressed in L. tarentolae, and because this system has recently begun to be sold commercially, it will be of interest to see what the future of this expression system is for producing challenging screening targets.
In vitro expression systems, based on E. coli transcription and translation machinery, have been used for many years to screen expression constructs for appropriate folding and function. 95 In vitro expression systems have not, however, been commonly used to produce protein targets for screening campaigns, mostly because it is much easier to scale up expression in E. coli than by using in vitro transcription and translation (IVTT) systems. A new cell-free mammalian IVTT expression system based on lysates from CHO cells may be able to provide some significant advantages to producing challenging mammalian proteins without the need to produce large volumes of mammalian cells in a sterile environment. 96 This system links transcription and translation, so the user can start with a DNA plasmid. Expression levels as high as 50 ug/ml have been attained. 96 Because this system uses CHO microsomes, both soluble and membrane proteins can be expressed. As with the L. tarentolae system, it will be interesting to see if the CHO IVTT system will be successfully used to produce and screen recombinant versions of challenging new targets.
Expression Systems for Membrane Targets
Membrane proteins, including the seven-transmembrane G-protein coupled receptors (GPCRs), represent the largest class of drug targets.11,97,98 Even though approximately 25% of all prokaryotic and eukaryotic genes encode membrane proteins, most still are considered proteins of unknown function, and only slightly more than 300 such proteins have high-resolution crystal structures. 99 It is clear from these observations that the overexpression of integral membrane proteins to sufficient levels to enable downstream purification and use in screening presents a unique set of challenges. These proteins are highly hydrophobic and prefer to exist in a lipid bilayer environment. Extraction of the membrane protein from the lipid bilayer environment of the cell wall using detergents often leads to loss of stability and activity. 100 Large numbers of experiments are often required to identify detergents that both efficiently extract the membrane protein from the cell wall and maintain its stability. 101 Experiments performed on the diacylglycerol kinase (DGK) enzyme revealed a dramatic effect on thermal stability and activity when the protein was solubilized in different detergents. DGK solubilized in dodecyl maltoside (DDM) retained >90% activity even when heated to 75°C but had <50% activity at 55°C when solubilized in lauryldimethylamine-N-oxide (LDAO). 102
Current high-throughput screening approaches for membrane protein targets often rely on cell-based assays or assays using isolated membranes. Both of these formats can be prone to artifacts because these systems contain not only the target of interest but also many other proteins that are present in the cell wall or peripherally associated with the membrane.103,104 Reporter-based cellular assays can reduce false-positive rates; however, it is important to select an assay that generates a biologically relevant readout, which is nontrivial.105,106 The ideal membrane protein-screening reagent would be one in which the target is pure, soluble, and active, which would necessitate the overexpression of a recombinant target protein to enable downstream purification. Mammalian membrane proteins are often subjected to posttranslational modifications, such as glycosylation, phosphorylation, lipidation, and ubiquitination, which would preclude their expression in a bacterial host system (e.g., E. coli).107–110 Baculovirus-infected insect cells, as discussed above, are capable of generating most, but not all, of the posttranslational modifications produced in mammalian cells, so the selection of the expression host will be highly dependent on how the protein is modified and what affect those modifications have on activity. 111 Expression of α1B-adrenergic receptor (AR) in different host cell lines demonstrated that the N-linked glycosylation and phosphorylation modifications were critical in maintaining structural heterogeneity of the receptor, whereas palmitoylation or O-linked glycosylation modifications were not. 112 Therefore, it may be suitable to express this receptor in baculovirus-infected insect cells because these cells produce N-linked glycosylations similar to those produced in mammalian cells. For other receptors in which O-linked glycosylation is essential for structural and functional fidelity, however, use of a mammalian host may be required. The baculovirus expression system has been used successfully for the generation of human GPCRs for structural studies, 113 but it is far from a panacea.114,115 A recent comparison of yeast and baculovirus expression systems for the production of human GPCRs demonstrated multiple advantages of yeast expression systems, including higher yields (in both the total protein and the fraction of active protein), ease of use, and shorter production times. 116 Use of the Bac-to-Mam system described previously for the infection of mammalian cells can produce transiently transfected cells displaying high levels of membrane protein expression, but the proprietary nature of this system limits its accessibility to small companies and academic research labs.117,118 Mammalian cells that have been adapted for serum-free growth in suspension at high density, such as Freestyle293 (Life Technologies) and Expi293 (Life Technologies), reduce the cell handling required for large-scale fermentations and transient transfections. Although these systems are advertised as being capable of producing milligram quantities of membrane proteins per liter of culture, in the authors’ experience the typical yields for a GPCR are often <1 mg/L of purified receptor.
Recent advances in determining the three-dimensional crystal structures of GPCRs have depended on approaches that require the modification of the protein, either by the insertion of a large, soluble fusion domain or by introducing multiple point mutations, both of which can alter the ligand-binding properties of the expressed receptor.119–121 The recently published crystal structure of glucagon receptor (GCGR) contains a thermally stabilized E. coli apo-cytochrome b562RIL (BRIL) domain in place of the native N-terminal domain of the receptor. 122 The N-terminal domain of the class-B GPCR plays a critical role in ligand binding and receptor activation, so it is unlikely that the GCGR–BRIL fusion construct would be capable of adopting a conformation competent for peptide binding. 123 Screening using highly modified constructs of membrane proteins would be more likely to produce false positives and reduce the likelihood of finding molecules with the desired biological activity. The structural determination of the first complex of a GPCR with its cognate G-protein was facilitated by the presence of a stabilizing immunoglobulin molecule (nanobody).124,125 It has been recently demonstrated that nanobodies can induce the formation of specific conformational states of the protein (i.e., agonist/activated). 124 The nanobody approach has the appeal of not requiring multiple mutations to the receptor itself, maintaining the conformation-specific ligand binding of the receptor and allowing for screening against a population of receptors locked into a defined conformational state. 126 The limitation of this approach is the ability to generate the nanobodies that require either isolation from the serum of inoculated llamas or screening against recombinant nanobody libraries. 127
Reconstitution of Membrane Targets
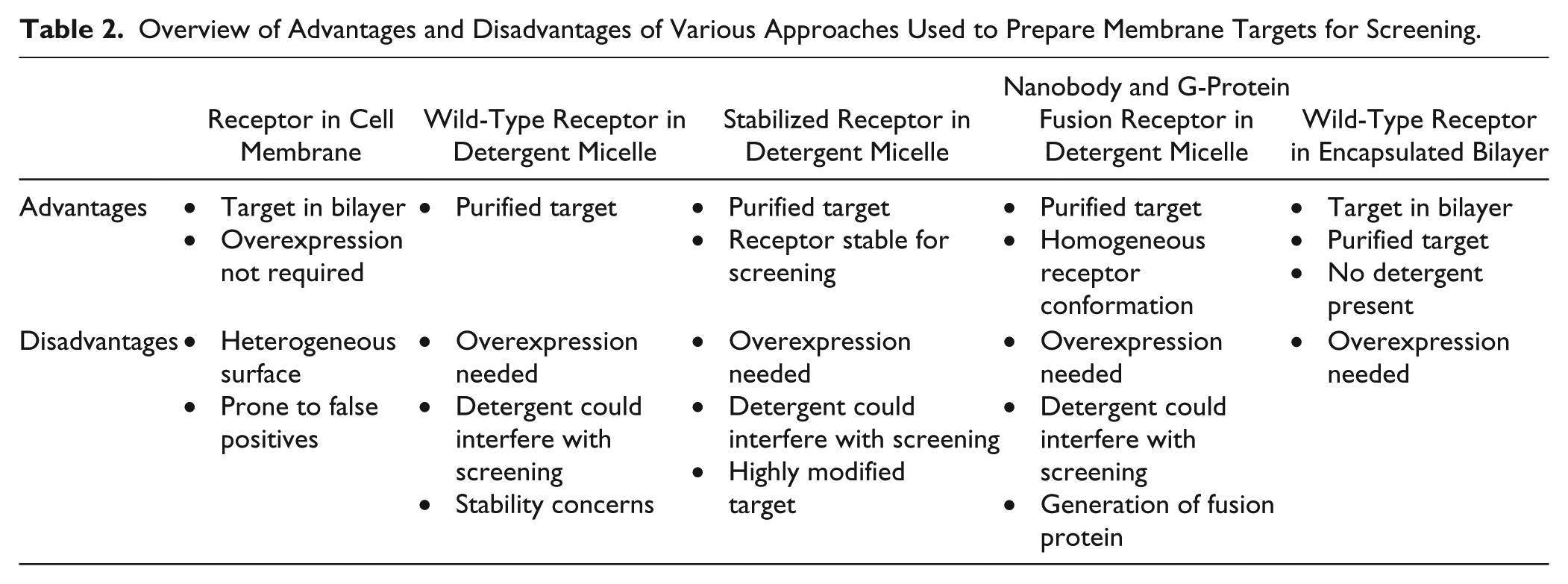
New approaches to reconstituting solubilized membrane protein targets in more native environments have focused on embedding membrane proteins in encapsulated lipid bilayers (nanodiscs) or solubilizing proteins in a mixed micelle, including lipids, detergents, and sphingolipids ( Table 2 ). The use of nanodiscs for this purpose is more broadly applicable, having been successfully applied to multiple membrane protein target classes, including GCPRs, cytochrome p450s, chemoreceptors, and many others, 128 and this approach does not require the mutation of the membrane protein itself. The nanodisc technique allows the membrane protein to be solubilized in the absence of detergent, which makes it appealing for applications that are sensitive to the presence of detergents.129,130 The nanodisc-embedded membrane protein is in a native, lipid-bilayer environment and is, therefore, more likely to adopt a biologically relevant conformation. The presence of lipids and other lipid-bound components can be critical for membrane protein function, as has been observed for gamma-secretase, a membrane-embedded protease involved in the proteolytic processing of the amyloid precursor protein (APP). The reconstitution of gamma-secretase into proteoliposomes of varying compositions demonstrated that the enzyme that had been reconstituted into proteoliposomes containing sphingolipids, cholesterol, and phospholipids had significantly increased activity relative to other preparations. 131
Overview of Advantages and Disadvantages of Various Approaches Used to Prepare Membrane Targets for Screening.
Discussion
So what is the best path forward to express a challenging new screening target? We have laid out a very simple flowchart to guide this process ( Fig. 1 ). Overall, the simplest path forward for a new target is to identify a homologous target that has already been successfully expressed and to follow the same method. It is mainly for this reason that there is such an extreme bias toward E. coli–expressed protein in both the structural and screening worlds. It has been estimated that only 15% of eukaryotic proteins can be expressed by E. coli in a soluble and active form, 132 and thus new expression systems are absolutely needed for novel targets. There have been many attempts to improve the E. coli system, with the addition of solubility-inducing fusion tags or the co-expression of multiple chaperones as described above, but sampling all of these methods with a new target is a tedious endeavor. Much more promising expression systems for the proper folding and posttranslational modification of target proteins, such as the multihost systems including mammalian cells, novel host organisms, and cell-free mammalian systems, have been developed in the past few years. Even production of some of the most challenging membrane targets in an environment very close to that of cellular membranes is becoming approachable with novel expression, purification, and reconstitution techniques. It will be of great interest to compare the results of screens conducted with protein produced from these novel systems to screening results and small-molecule drug-development campaigns with E. coli–expressed targets. It will also be of interest to assess how many novel targets will become accessible to screening campaigns as a result of these innovative technologies. The ultimate question is whether these new expression methods will produce more efficacious small molecules either by accessing novel targets for disease treatments or by producing screening reagents that more faithfully represent the endogenous targets. Perhaps the former is more likely than the latter, but in both cases, the production of challenging new targets by novel expression systems significantly increases the likelihood of identifying the exciting new small-molecule therapies of the future.
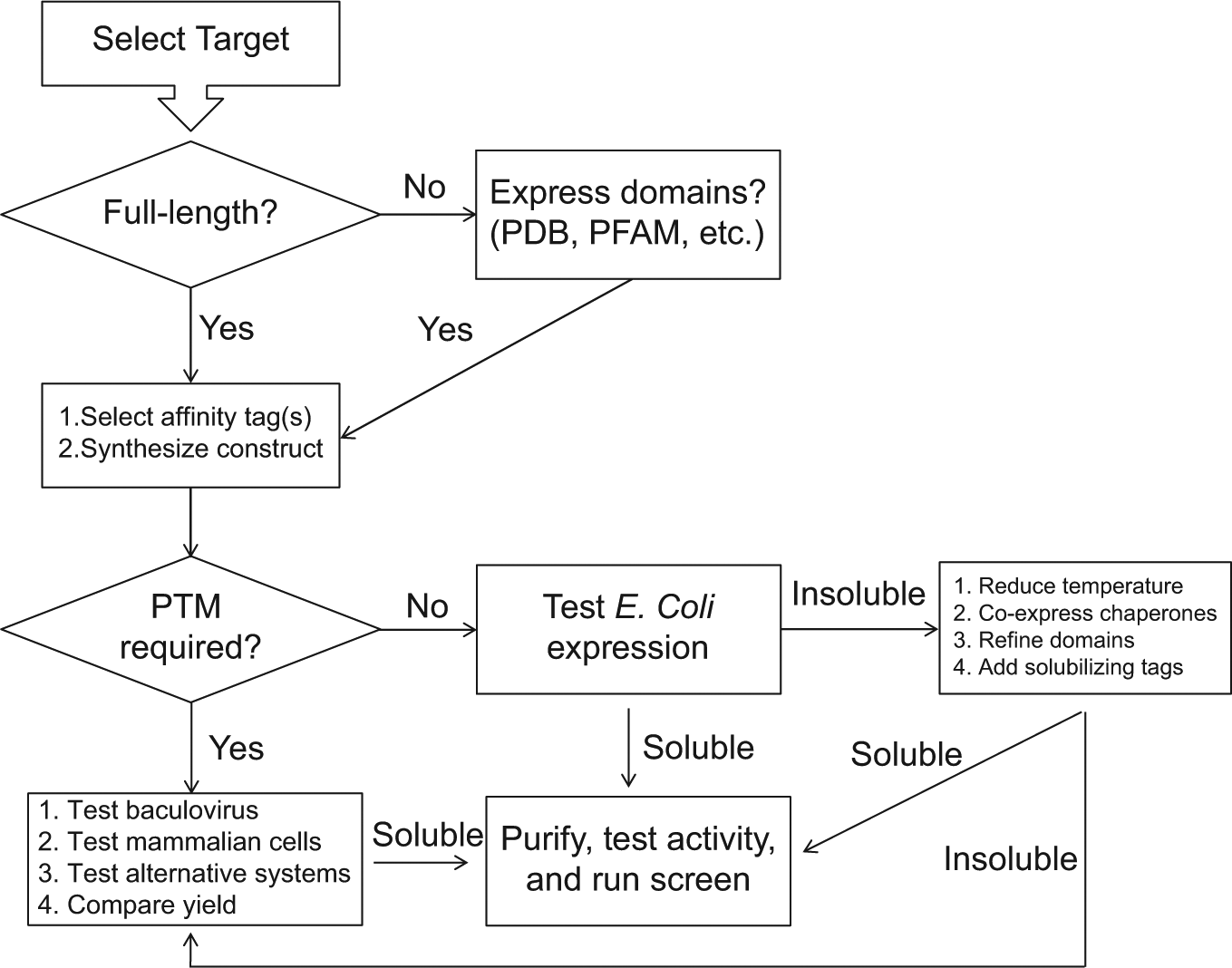
Simplified flowchart for protein expression. PDB, Protein Data Bank (http://www.pdb.org); PFAM, Protein Families (http://pfam.sanger.ac.uk); PTM, posttranslational modification.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.