
Abstract
Three-dimensional (3D) cell culture models are gaining increasing interest for use in drug development pipelines due to their closer resemblance to human tissues. Hydrogels are the first-choice class of materials to recreate in vitro the 3D extra-cellular matrix (ECM) environment, important in studying cell-ECM interactions and 3D cellular organization and leading to physiologically relevant in vitro tissue models. Here we propose a novel hydrogel platform consisting of a 96-well plate containing pre-cast synthetic PEG-based hydrogels for the simple establishment of 3D (co-)culture systems without the need for the standard encapsulation method. The in-depth density gradient at the surface of the hydrogel promotes the infiltration of cells deposited on top of it. The ability to decouple hydrogel production and cell seeding is intended to simplify the use of hydrogel-based platforms and thus increase their accessibility. Using this platform, we established 3D cultures relevant for studying stem cell differentiation, angiogenesis, and neural and cancer models.
Introduction
Three-dimensional (3D) cell culture platforms represent a better mimic of the biological milieu compared with conventional cultures on flat (2D) rigid substrates. The use of such platforms in the study of biological mechanisms and for phenotypic screenings in drug discovery is growing. 1
Hydrogels are the first-choice class of materials for re-creating in vitro the 3D extracellular matrix (ECM) environment, important for the study of cell-ECM interactions and 3D cellular organization, leading to physiologically relevant in vitro tissue models. 2 The encapsulation method is the standard method for forming 3D cell-laden hydrogel constructs. This method consists of mixing cells with a liquid hydrogel precursor and waiting for the mixture to polymerize via a covalent cross-linking reaction or via a noncovalent physical polymerization. 3 The encapsulation process can be carried out using robotic liquid-handling devices to achieve reproducibility and compatibility with microtiter plate formats.4,5 Nevertheless, mastering the complex chemical and operational process parameters is necessary to achieve this end. Additional requirements are dedicated equipment and often preparative steps, such as well-plate pretreatment 5 and equipment precooling. 6 These requisites limit the adoption of innovative hydrogel platforms for assay development in academia and industry. Ready-to-use solutions could make hydrogels more accessible to users lacking the expertise in hydrogel chemistries and support the development of hydrogel-based assays. Such platforms require the separation of the hydrogel production and cell seeding.
Decoupling hydrogel formation and cell addition is hindered by the fact that most cell types seeded on the surface of preformed hydrogels typically grow in a monolayer at the hydrogel surface and do not—or poorly—invade the 3D environment. 7 To overcome this problem, researchers have developed a number of strategies to improve hydrogel permeability to cells, including the creation of macropores 8 and the production of chemical 7 or physical gradients 9 spanning from the hydrogel surface to the bulk. Capitalizing on these technologies for promoting cell infiltration enables the development of ready-to-use hydrogel platforms.
Here, we adapted a recently developed manufacturing method for the production of fully synthetic poly(ethylene glycol) (PEG)–based hydrogels featuring an in-depth cross-linking density gradient 9 to the microtiter plate format (3DProSeed 96-well plate). We previously showed that the in-depth cross-linking density gradient promoted the infiltration in 3D of mesenchymal stem cells (MSCs) seeded at the hydrogel surface. 9 The objective of this work was to assess the versatility of this platform for a number of 3D cell cultures produced via cell infiltration through the hydrogel’s engineered surface. To this end, we seeded various cell types (and some combinations) including mesenchymal stem cells, endothelial cells (ECs), neuronal cells, and epithelial cancer cells on the hydrogel featuring the in-depth surface density gradient and investigated their ability to penetrate the gel in 3D and establish culture models morphologically similar to the conventional systems based on encapsulation.
Materials and Methods
Preparation of Hydrogel-Gradients Plates
We produced gradient hydrogels based on previously established transglutaminase–cross-linked PEG hydrogels, that are RGD functionalized and matrix metalloprotease (MMP) sensitive. 10 In brief, hydrogels with a final dry mass content of 1.7% were prepared by FXIIIa (thrombin-activated factor XIII, 10U/mL)-mediated crosslinking of precursor solution, containing a stoichiometrically-balanced mixture of PEG monomers functionalized with glutamine acceptor peptide substrates and lysine donor peptide substrates. The peptide substrates contain an MMP-sensitive and RGD motives. The precursor solutions were prepared in Tris buffer (pH 7.4) with 50mM calcium chloride. The gel stiffness was chosen according to previously optimized cross-linking density for 3D mesenchymal cell migration by encapsulation. 11
By locally inhibiting the polymerization of the upper surface of the gel using electrochemical processes, we produced hydrogels featuring an in-depth cross-linking gradient. 12 The gradients span more than 400 µm from the surface (minimal density) to the bulk (maximal density), as obtained in our previous work by applying 1 µA per square millimeter. 9 This process was made compatible with the fabrication in microtiter plates by multiplexing simultaneously in all 96 wells ( Fig. 1A ).
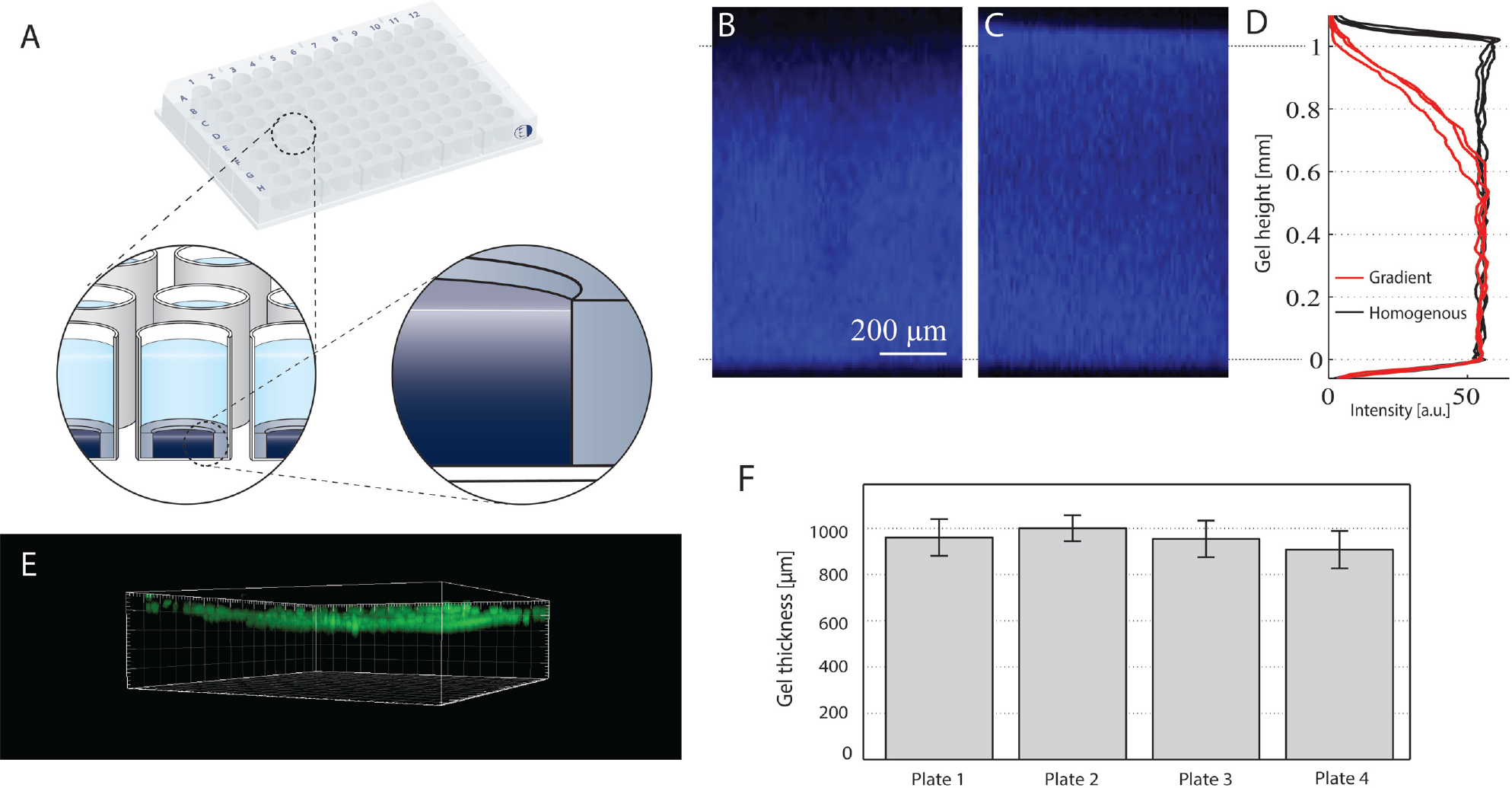
Hydrogel platform characterization. The hydrogel 96-well plate schematized in (
Assessment of Hydrogel Cross-Linking Density
By admixing fluorescein isothiocyanate (FITC)–tagged Lys substrates (Lys-FITC) with the hydrogel precursors, the fluorescent molecule is cross-linked within the hydrogel following the same polymerization mechanism. 9 The incorporation of FITC in the gel is directly correlated to the cross-linking density, and its intensity can be used to evaluate cross-linking efficiency. Using confocal laser scanning microscopy (CLSM; SP5, Leica, Germany), stacks of 20 µm slice increments were acquired through the entire thickness of the gel for mapping the cross-linking density in the z-direction. To account for the loss of laser power through the sample, signal compensation was applied following a calibration performed in a homogeneous sample, as described by the instrument’s manufacturer. To obtain the intensity profiles, the minimum intensity (zero) was set using the signal intensity above the hydrogel surface. The same parameters were used for all samples.
Evaluation of Intraplate Reproducibility
To evaluate the well-to-well and plate-to-plate reproducibility of the process, we measured the hydrogel height. For this, 20 μm fluorescent beads (Polyscience, Fluoresbrite YG 190962) were dispensed and left to settle on the hydrogel surface. An inverted microscope with controlled stage (Leica DMI6000) was used to determine the distance between the bottom of the well and the focal plane of the beads. Twelve wells of four different well plates were measured to determine the intraplate variation (average and standard deviation within a plate).
Cell Culture Material
A list of used cells and media components is provided in Table 1 .
List of Cells and Media and Respective Providers Used Throughout This Work.
Cell Invasion
To assess the ability of cells to migrate into hydrogel interfaces, we seeded 5.0 × 103 to 4.0 × 104 MSCs on top of the engineered gel surfaces and inspected the cell distribution over the uppermost 500 µm of the hydrogel after 1, 3, 7, and 10 d in culture using CLSM, as described in our previous work. 9 At every endpoint, samples were fixed with 4 % paraformaldehyde. After stopping the reaction with 0.1 M glycine in phosphate-buffered saline (PBS; pH 7.2), samples were washed twice with PBS. Permeabilization was performed for 20 min at room temperature with 0.2% Triton X100 in PBS followed by two washing steps with PBS. Then cells were stained for f-actin incubating overnight at 4 °C with rhodamine-labeled phalloidin (1:250 in PBS; Life Technologies, Carlsbad, CA) and rinsed three times in 1 h with PBS.
Platform Versatility
Epithelial Cyst Formation
A total of 1.0 × 104 MCF10A cells were seeded and cultured for 10 days on homogeneous and gradient gels.
Samples were fixed and stained for f-actin (as described above) and for cell nuclei using Hoechst stain (1:1000 in PBS for 30 min; Life Technologies, Cat. No. H3570). Subsequently, samples were again washed three times with PBS. Cell invasion and formation of acinar structures were assessed by CLSM.
Pancreatic Carcinoma Tumoroid Formation
A total of 1.0 × 104 PaCa cells were seeded and cultured for 15 d on homogeneous and gradient gels. Samples were fixed and stained for cytokeratin 19 (CK19). After fixation and permeabilization (as described above), CK19 was stained over night at 4 °C with mouse anti-CK19 antibody (1:200 in PBS; Cat. No. ab9221, Abcam, Cambridge, UK), followed by rinsing three times in 1 h with PBS and a 4-h incubation with an Alexa Fluor 488 goat anti-mouse IgG (1:200 in PBS; Abcam, Cat. No., ab150113). Cell invasion and formation of cell spheroids were assessed by CLSM.
Neural Culture and Neurite Outgrowth
A total of 4.0 × 104 iPS-derived neurons (CNS.4U, Axiogenesis, Köln, Germany) were seeded and cultured for 15 d on homogeneous and gradient gels. Samples were fixed and stained for tubulin (TUJ1, 1:300 in 3% bovine serum albumin in PBS; Cat. No. T2200, Sigma-Aldrich, St. Louis, MO). TUJ1 was stained overnight at 4 °C and rinsed three times in 1 h with PBS followed by a 4-hour incubation with an Alexa Fluor 488 goat anti-rabbit IgG (1:200 in PBS; Abcam, Cat. No. ab150113). The cell invasion and neurite outgrowth were assessed by CLSM.
Calcium Imaging
To obtain an optical readout of intracellular calcium fluctuations indicative of neural activity, neurons were transduced with a neuron-specific AAV GCaMP6 vector (AAV1.Syn.GCaMP6f.WPRE.SV40; Penn Vector Core, University of Pennsylvania, Philadelphia, PA) after 10 d in vitro at a 1:1000 dilution by volume. Culture medium was fully replaced 5 d after the addition of the viral vector. Neurons were imaged at 17 d in vitro with an epifluorescent microscope that acquired an image every 100 ms to assess the transient calcium.
Endothelial Cell Co-Cultures
A total of 1.0 × 104 green fluorescent protein–labeled human umbilical vein endothelial cells were co-seeded with 1.0 × 104 MSCs and co-cultured for 7 d in a 75% MSC medium–25% EC medium mixture. At the endpoint, samples were fixed and stained for f-actin. Cell invasion and hollow tubelike structure formation were observed by CLSM.
To access the angiogenic potential of growth factors, 50 ng/mL fibroblast growth factor (FGF), 50 ng/mL vascular endothelial growth factor (VEGF), or 50 ng/mL Netrin 4 was added to the culture medium. After 1 week of co-culture, the samples were imaged with an epifluorescence microscope (Leica, DMI600), and the average lengths of individual microvascular structures formed by the green fluorescent protein (GFP)–expressing endothelial cells were quantified with ImageJ for three independent samples per condition. All mean values were compared by one-way analysis of variance using Matlab 7.9 (MathWorks Inc., Torrance, CA). Statistical significance was accepted for p < 0.05 after comparing the mean values by Bonferroni post hoc test.
Sequential Seeding
To confirm that cells seeded at different time points can invade the gradient gels and not the homogeneous gels, 104 MSCs were seeded on gradient gels and cultured in MSC medium. After 7 d of culture, 104 red fluorescent protein–labeled fibroblasts (RFP-HFFs) were added on top of these precultured gels and left in culture for another 7 d. As a control, MSCs were encapsulated in homogeneous gels (1 × 106 cells/mL gel) and left in culture for 7 d prior to adding RFP-HFFs for another 7 d. Samples were then fixed and stained with Alexa 633-labeled phalloidin. The co-culture was then assessed by CLSM.
To study the interaction of pancreatic cancer cells with a fibroblast-rich environment, 104 RFP-HFFs were seeded on gradient gels and cultured for 7 d; 104 PaCa cells were sequentially added to the culture and co-cultured for up to 15 d.
At different time points during the 3 to 15 d of co-culture, samples were fixed and PaCa cells stained for CK19 and counterstained with Hoechst stain for nuclei (as previously described). Co-cultures were then assessed by confocal or epifluorescent microscopy.
Results and Discussion
Description and Characterization of the Hydrogel Plate
We produced 96-well plates containing gels featuring an in-depth surface density gradient (referred to as gradient gels, as opposed to homogeneous gels featuring a homogeneous density throughout the gel). The hydrogel is PEG based, fully transparent, and made permissive to cell migration via the incorporation of MMP-cleavable sites. Figure 1A shows the schematic of the 96-well plate with a cross section of a well. The hydrogel is formed within an inner well (4 mm diameter), the edge of which facilitates manual handling that could otherwise result in damage to the gel due to the gel’s transparency. Figure 1B and C are X-Z projections of a FITC-labeled gradient gel and homogeneous gel, respectively, acquired with CLSM. By covalently labeling the hydrogel with FITC, we used the fluorescence signal to assess the change of cross-linking density over the z-axis. The surface gradient typically extended 400 µm from the surface ( Fig. 1B , D ), whereas homogeneous gels do not feature any gradient ( Fig. 1C , D ). To assess the reproducibility of the hydrogel thickness, we deposited fluorescent beads on top of the gradient gels ( Fig. 1E ) and measured the height from the glass bottom. For four plates, the mean gel thickness and standard deviation are shown in Figure 1F .
Culture and Differentiation of MSCs
MSCs seeded on the gradient gels immediately adopted a typical 3D phenotype characterized by a spindlelike shape ( Fig. 2A–C ).
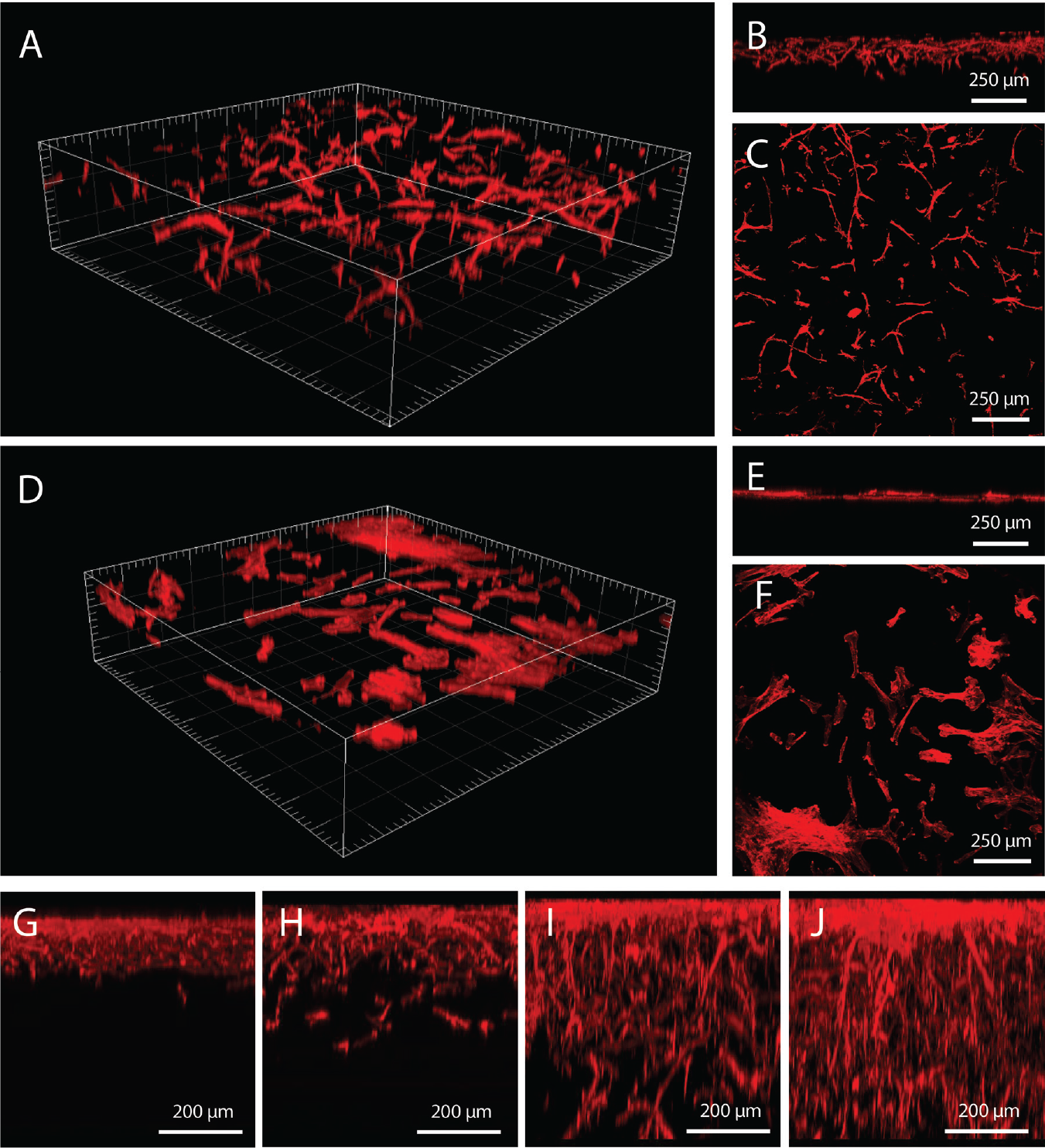
Mesenchymal stem cell (MSC) behavior on hydrogel gradients. On each gradient gel (
MSCs invaded the gradient gels over time, reaching 200 μm within 3 days after seeding and populating the gel to more than 500 μm in depth after 10 days of culture (
Fig. 2G–J
). MSCs are able to migrate through regions of the gel featuring the highest density, indicated by the plateau in
Figure 1D
and referred to as bulk density. In fact, MSCs encapsulated in hydrogels made of the bulk density were able to migrate in 3D.9,11 Nevertheless, MSCs seeded on the homogeneous gels grew on the gel surface in a monolayer (
Fig. 2D–F
), and did not penetrate the hydrogel after 10 d of culture (
To confirm that MSCs keep their pluripotency during the invasion of the gradient gels, MSCs were allowed to infiltrate the hydrogel for 7 d and were differentiated toward adipogenic and osteogenic phenotypes. After 15 d of adipogenic differentiation, we observed increased amounts of red oil–stained lipid droplets in cultures exposed to inducing medium compared with control cultures (
Cells modulate their environment by secretion and deposition of their own ECM. To confirm the presence of endogenously deposited fibronectin and collagen, we used specific immunostainings (
Versatility of the Platform in Terms of Cultured Cells
In this section, we compared the behavior on homogeneous and gradient gels of other cell types, relevant for screening applications in oncology, neurotoxicology, and angiogenesis.
Breast Epithelial Cells
The MCF10A is a commonly employed nontumorigenic breast epithelial cell line, which leads to the formation of acinar structures when encapsulated in Matrigel or other hydrogels. Acinar structures consist of polarized and organized cell spheroids featuring a lumen and are used to model mammary gland development, 16 tumorigenesis, and the potential of molecules to reverse this process. 17 When cultured in 2D, also on top of homogeneous gels ( Fig. 3A ), MCF10A cells formed a dense monolayer. In contrast, MCF10A cells seeded on the gradient gels infiltrated homogeneously in 3D and started forming acinar structures within 3 d of culture ( Fig. 3B–D ). Similarly to when encapsulated in hydrogels of the same formulation, only a minor fraction of about 10% of the cells developed into acinar structures. This fraction could be increased by adding defined fragments of proteins. 18
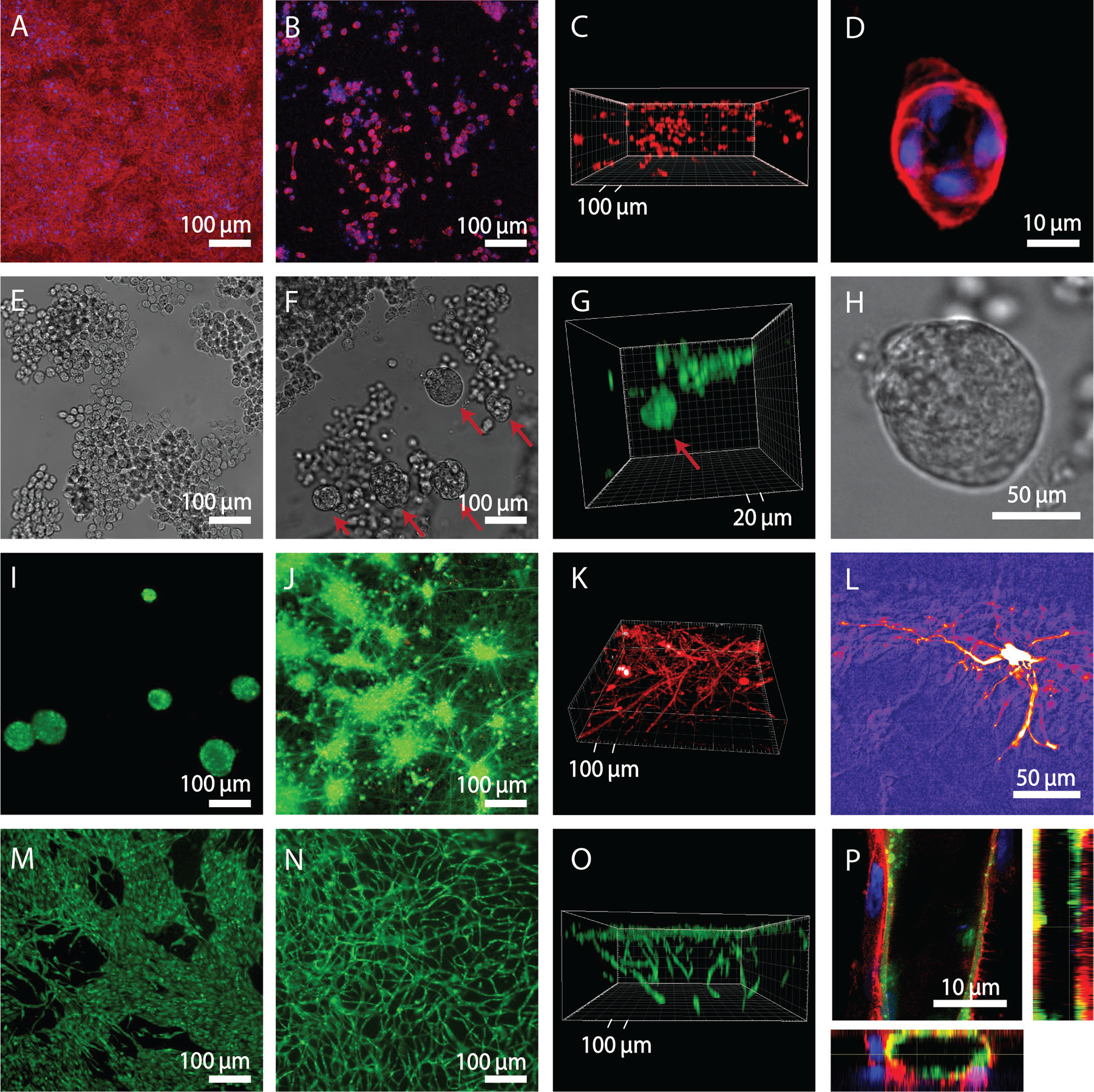
Versatility of the platform. MCF10A epithelial cell (10 d of culture,
Carcinoma Cells
Pancreatic cancer cells form monolayers when cultivated on flat surfaces. However, when encapsulated in 3D hydrogels, carcinoma cells form spheroids similar to tumor masses found in patients. These 3D tumor models exhibit more physiologically relevant protein expression profiles and responses to anticancer drugs compared with 2D cultures.19,20
On homogeneous gels, pancreatic carcinoma cells grew on the surface as a monolayer ( Fig. 3E ). In contrast, on gradient gels, some cells were able to penetrate and form spheroids developing into 100 µm structures after 15 d of culture ( Fig. 3F–H ). Sieh et al. 20 cultured prostate carcinoma cells encapsulated in similar hydrogels and observed after 28 d the formation of spheroids growing to 200 µm in diameter and featuring apoptotic cores similar to the in vivo situation.
Neurons
Hydrogel-based 3D culture conditions have been shown to enhance the maturation of neural cultures compared with 2D, especially for modeling Alzheimer’s disease.
21
Human induced pluripotent stem cell (iPSC)–derived neurons formed spherical clusters when seeded on the homogeneous gels (
Fig. 3I
), which appeared similar to those forming on any nonadhesive surface. These clusters were viable over 3 wk, but no protrusion of cell bodies and neurites into the gel was observed. In contrast, when seeded on gradient gels, iPSC-derived neurons extended the neurites throughout the hydrogel (
Fig. 3J
,
K
) and formed functional networks generating spontaneous activity, as indicated by calcium imaging (
Figure 3L
;
Angiogenesis
Regulating angiogenesis is critical for tumor growth and metastasis and represents an important strategy to fight tumor progression. 23 Consequently, a number of in vitro angiogenesis models have been developed for screening applications. 24 ECs lining the lumen of blood vessels have been shown to be centrally involved in the formation of new vessels by processes such as sprouting and intussusception. 25 The encapsulation in hydrogels of ECs together with supporting cells leads to the formation of 3D networks resembling native capillaries, which connect to host vasculature of mice when implanted subcutaneously. 14 For ECs co-seeded with supporting cells on the gradient gels, ECs migrated inside the gel and assembled in 3D tubelike structures ( Fig. 3N–P ). These structures consist of networks of endothelial cells forming hollow structures surrounded by support mesenchymal cells. Without the supporting cells, ECs alone migrated into the gels and started elongating but failed to establish stable structures (not shown here), as described when encapsulated in similar gels. 26 On the homogeneous gels, ECs and support cells grew in a monolayer, each segregated from the other ( Fig. 3M ). Only after a prolonged culture time (approximately 2 wk) did the cells form cordlike structures (not shown here) similar to those previously observed in 2D cultures. 27
We tested the proangiogenic effect of a small panel of compounds on the prevascular structures created by seeding ECs and supporting cells on the gradient gels and can confirm the results previously obtained on other 2D or 3D culture systems. We used the fluorescence signal of GFP-ECs to qualitatively ( Fig. 4A–D ) and quantitatively ( Fig. 4E ) assess the endothelial cord length following addition of FGF, VEGF, and Netrin 4. FGF significantly increased the mean cord length compared with all the other factors, whereas VEGF significantly increased the mean cord length compared with the control and Netrin 4. These results are in accordance with the literature stating that FGF and VEGF are potent proangiogenic factors, and FGF is the strongest promoter of microvessel formation. 28 This proof-of-concept compound testing demonstrates the feasibility of performing 3D angiogenesis screenings on ready-to-use hydrogels, without the need of encapsulation.
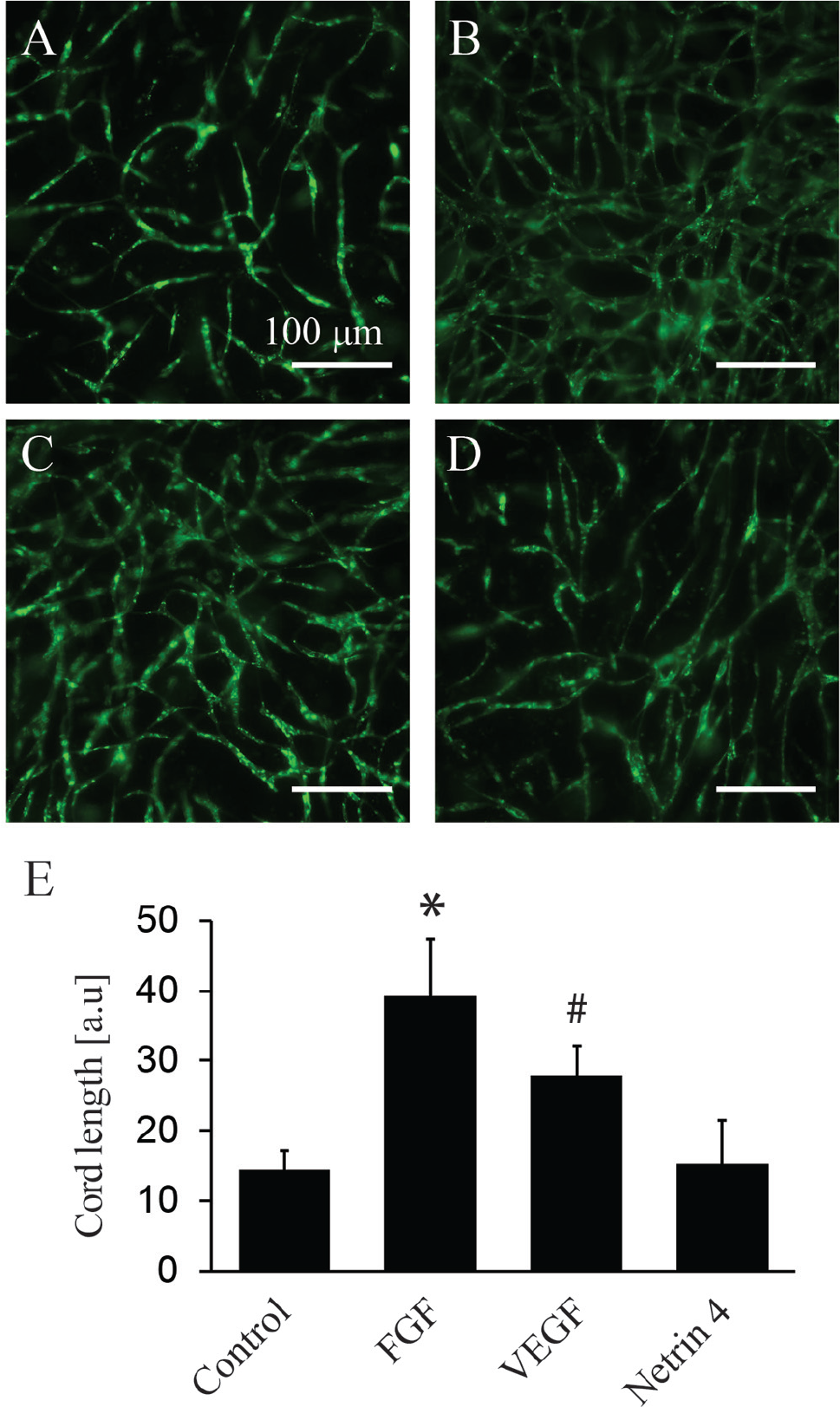
Compound testing angiogenesis assay. Co-cultures of green fluorescent protein (GFP)–endothelial cells (green) and mesenchymal cells (not stained) were cultured for 7 d without additional growth factor: control (
Sequential Seeding of Different Cell Populations
Advanced cell assays often require the addition of different cell types at different time points to create co-culture systems. First, cells are cultured to create a microenvironment, to which other cells of interest can be added, and a test is carried out. These co-culture assays are valuable in the field of oncology, as the tumor microenvironment and surrounding stromal tissue affect the tumor fate and response to therapies. For instance, Ghajar et al. 29 showed how the perivascular niche regulates the dormancy or proliferation of breast cancer cells by sequentially adding cancer cells to a pre-established 2D vasculature.
Impressive co-cultures have been established in hydrogels via co-encapsulation of different cell populations. George et al. 30 recapitulated the extravasion of specific cancer cells from a microtissue in the surrounding fibrin hydrogel through microvessels. In such models, the scientists could not uncouple the formation of the microenvironment and the tumor growth.30–32 Because cells have significant differences in proliferation rate, the most proliferative types tend to overgrow the others, and adding cells at a different stages of the culture would be beneficial. 33 However, sequential seeding requires the newly added cells to be able to penetrate and interact with the pre-established microenvironment (depicted in Fig. 5A ).
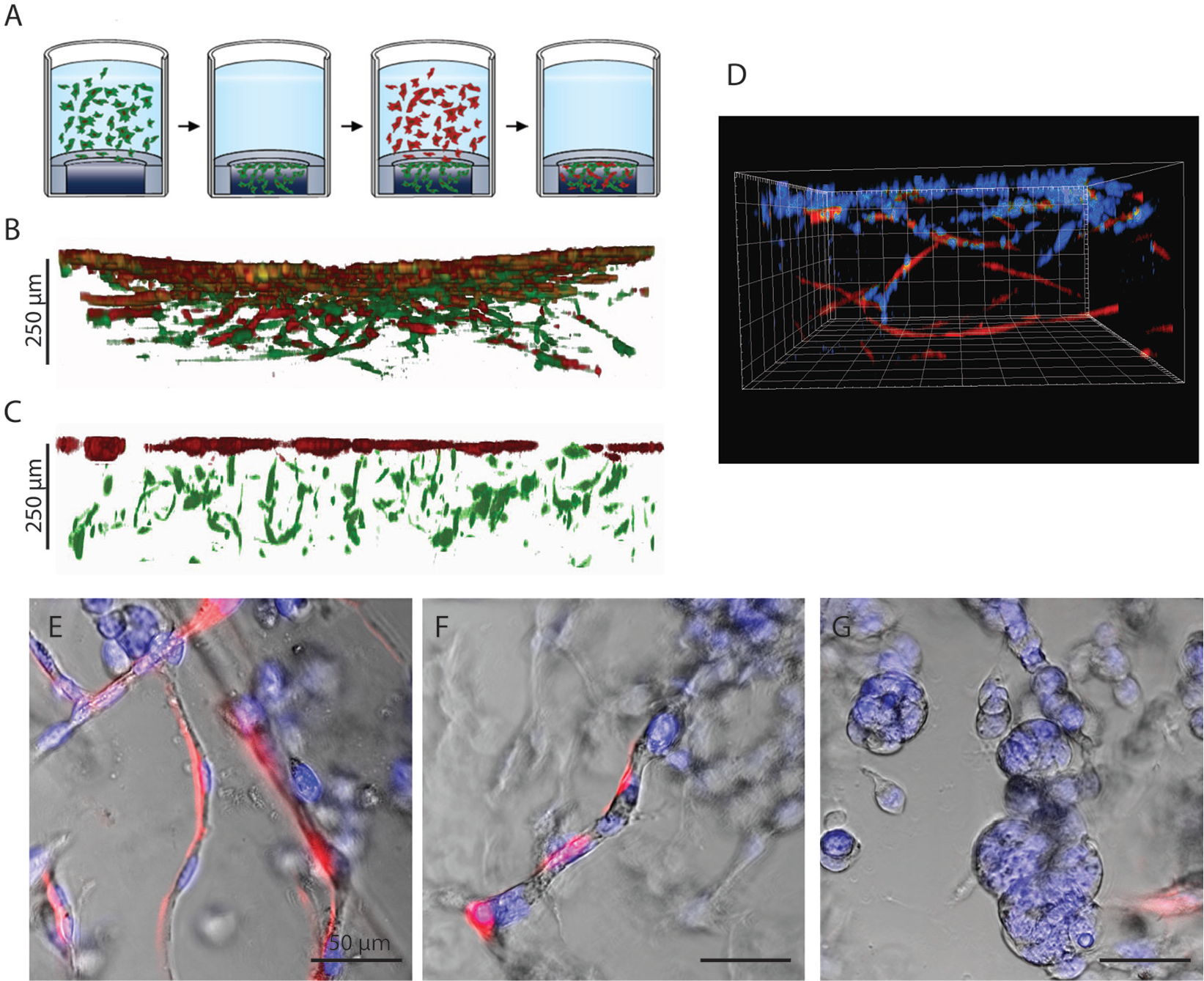
Sequential seeding. Gradient gels allow for the sequential addition of cells at different time points, as depicted in schematic
Here, we added a second population of fibroblasts labeled with RFP to a gradient gel preseeded with a first population of MSCs labeled with GFP and observed that the RFP fibroblasts migrated into the gel and established a co-culture in 3D ( Fig. 5B ). In contrast, when the second fibroblast population was seeded onto an MSC-laden gel made by encapsulation, the second fibroblast population remained confined at the surface and did not penetrate into the hydrogel ( Fig. 5C ). We therefore believe that the gradient gels offer an elegant alternative for adding to hydrogel different cell populations at selected time points during an assay.
Cancer-associated fibroblasts play a great role in modulating cancer invasion. 34 Researchers have shown that fibroblasts help cancer invasion through hydrogels, either actively or by creating tracks followed by the cancer cells.35,36 Here, we seeded pancreatic carcinoma cells on a gradient gel preseeded with fibroblasts and cultured them for 2 wk. Cancer cells migrated inside the gel by crawling along the fibroblasts, and once in the gel, they started forming microtissues ( Fig. 5D–G ).
This platform can be used to seed different cell populations into a hydrogel at different points in time, to establish co-culture systems without the need for encapsulation. In the future, this platform could be used to study the growth of tumor cells in a range of microenvironments with controlled ECM deposition, tissue types, or even prevascularized constructs.
In conclusion, the presented hydrogel platform, featuring an in-depth surface density gradient, enables the simple establishment of 3D hydrogel-based cultures by seeding cells similarly to that conventionally done on standard 2D plates. Cells seeded onto gradient hydrogels can mature and assemble into 3D structures comparable with the ones created via encapsulation. Moreover, gradient hydrogels can be used to sequentially seed different cell populations to create 3D co-culture systems. These data demonstrate the use of this platform for a variety of cell types and tissue models, but the applicability to other cell types not presented here needs further investigation.
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: V.M. and B.R.S. are employed by Ectica Technologies AG, a company developing the 3DProSeed 96-well plate, which may be considered a conflict of interest to this work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Swiss National Science Foundation (SNSF), the Centre for Clinical Research University Hospital and University of Zurich, ETH Zurich, the National Science Foundation of China (No. 31671008) and Chinese Scholarship Council and the NCCR Molecular Systems Engineering.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.