
Abstract
Cells grown in 3D are more physiologically relevant than cells cultured in 2D. To use 3D models in substance testing and regenerative medicine, reproducibility and standardization are important. Bioprinting offers not only automated standardizable processes but also the production of complex tissue-like structures in an additive manner. We developed an all-in-one bioprinting solution to produce soft tissue models. The holistic approach included (1) a bioprinter in a sterile environment, (2) a light-induced bioink polymerization unit, (3) a user-friendly software, (4) the capability to print in standard labware for high-throughput screening, (5) cell-compatible inkjet-based printheads, (6) a cell-compatible ready-to-use BioInk, and (7) standard operating procedures. In a proof-of-concept study, skin as a reference soft tissue model was printed. To produce dermal equivalents, primary human dermal fibroblasts were printed in alternating layers with BioInk and cultured for up to 7 weeks. During long-term cultures, the models were remodeled and fully populated with viable and spreaded fibroblasts. Primary human dermal keratinocytes were seeded on top of dermal equivalents, and epidermis-like structures were formed as verified with hematoxylin and eosin staining and immunostaining. However, a fully stratified epidermis was not achieved. Nevertheless, this is one of the first reports of an integrative bioprinting strategy for industrial routine application.
Introduction
Three-dimensional (3D) cell culture technology has undergone rapid development as an alternative to traditional 2D cell cultures. 1 In vitro 3D culture systems promise more reliable and predictive tissue-like models for drug development and substance testing.2–4 This has been emphasized in the recently published special theme issue of Advanced Drug Delivery Reviews (“Innovative Tissue Models for Drug Discovery and Development,” http://www.sciencedirect.com/science/journal/0169409X/69-70).
Organ-like tissue models need to exhibit a certain degree of complexity to reflect the in vivo situation as closely as possible. With respect to this highly demanded complexity, bioprinting shows a great potential to produce artificial 3D tissues and organs.5–6 The advantage of this technology compared to standard tissue-engineering approaches is the exact positioning of cells and bioactives, such as signaling factors and matrix components (so-called bioinks), to obtain spatial control. 7
To print 3D soft tissue–like structures, cells are either mixed with bioink and printed or printed separately as cell suspensions onto bioink. The 3D construct is assembled in an additive manner by printing layer by layer. Three different approaches are pursued for the printing of living tissue models: (1) laser-assisted bioprinting (LaBP), (2) inkjet printing, and (3) robotic dispensing (reviewed in Refs. 8–9). Besides the bioprinting instruments’ properties, the characteristics of the bioinks are of prime importance to obtain high-fidelity printed constructs. Therefore, much effort is actually been put into the development and characterization of bioinks that not only have to be printable and viscous enough to maintain the structure before polymerization but also have to provide the perfect extracellular matrix (ECM)-like environment.9–10 Several naturally derived and synthetic hydrogel materials are commonly used as bioinks, such as alginate, 11 gelatin, 12 collagen I, 13 matrigel, 14 fibrinogen, 15 hyaluronic acid, 16 agarose, 17 chitosan, 18 and poly(ethylene) glycol (PEG). 19 All of these polymers show distinct polymerization modes to produce stable scaffolds in an additive manner. 9 But, until today, no hydrogel has been developed and commercialized for standardized bioprinting processes.
In Europe, since July 2013, it is mandatory by law to assess the potential hazard of cosmetic ingredients with in vitro skin models because animal tests are banned.20–21 Therefore, artificial skin is one of the best-characterized tissue models, and several validated models are currently commercially available. Also in drug development, these models are expected to substantially contribute to assessing systemic drug application on the skin and the effect of drugs to treat skin diseases or disorders. 22 Bioprinting technology is currently under investigation to produce in vitro skin to standardize the manufacturing process. Lee et al., 23 in a primary study, showed the feasibility to print defined layers of human primary fibroblasts and keratinocytes in a collagenous matrix using the inkjet-based printing approach. The collagen I precursor solution was jetted and solidified on a pH change after addition of sodium bicarbonate by a nebulizer. In a follow-up study, Lee and coworkers 24 optimized the skin bioprinting approach using the human cell lines foreskin fibroblasts-1 (hFF-1) and keratinocytes (HaCaT). After culture of the HaCaTs at the air–liquid interface, the cells started to differentiate and to express epidermis-specific proteins, such as N-cadherins. However, a fully stratified epidermis could not be achieved with cell lines.
Koch et al. 25 used LaBP to build skin-like structures with cell lines from mouse fibroblasts (NIH3T3) and HaCaTs embedded in a collagen matrix transferred to a matrigel support. The models were characterized with histological analyses for the expression of several skin-specific proteins such as pan-cadherin, connexins, and laminin. In a subsequent study by Michael et al., 26 the printed skin construct was transplanted into a dorsal skinfold chamber in nude mice and fully integrated into the surrounding tissue. However, the usage of cell lines instead of primary cells was the main limitation of this study.
In this report, we developed an all-in-one-solution for a reproducible bioprinting process, including customized solutions of a ready-to-use printable matrix (BioInk) combined with the commercialized bioprinter 3DDiscovery and a printing protocol. In a proof-of-concept study, a skin model was developed as a soft tissue reference. Primary human dermal fibroblasts were printed onto BioInk layers. The printed dermis equivalents were long-term cultured, showing cell viability and proliferation as well as the capability to remodel the matrix as verified by collagen type I expression. Moreover, the dermal models supported primary keratinocyte differentiation. Printed skin equivalents were characterized histologically and demonstrated the feasibility to print skin-like constructs with two primary cell types.
Our integrative bioprinting strategy should contribute not only to standardizing the bioprinting process but also to producing tissue models on demand in a reproducible way for routine industrial application.
Materials and Methods
Cell Expansion
Human primary dermal fibroblasts (HDFa; Cascade Biologics, Invitrogen, Basel, Switzerland) were cultured in monolayer using Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Gibco, Waltham, MA) (HDFa medium I). Human primary epidermal keratinocytes (HPEK; CELLnTEC Advanced Cell Systems, Bern, Switzerland) were cultured in CnT-57 proliferation medium (CELLnTEC) at 37 °C and 5% CO2. Media were exchanged every 3 days, and the cells were subcultured at 70–80% confluency. For the printing experiments, HDFas were used up to passage 6, and HPEKs up to passage 5.
Bioprinter
The bioprinting device 3DDiscovery ( Fig. 1 ) consists of an x-y-z-axis positioning system with a tool charger equipped with four work stations and a building platform. The tool charger moves in the x- and z-axes. The building platform, at which cell culture vessels (e.g., well plates and inserts) are placed, moves in the y-direction. Each work station is equipped with an independently controllable printhead. Biomaterial (e.g., ECM molecules, cells, and signal molecules) is provided in 3 ml disposable cartridges fixed onto the printhead with luer lock connections. Liquid dispensing with the cell-compatible printheads is controlled by electromagnetic microvalves based on inkjet technology and sterile compressed air with an accuracy of ±10 nl. To print under temperature-controlled conditions, a cartridge-cooling unit is integrated into the bioprinter. An ultraviolet (UV) light-emitting diode (LED) unit (λ = 365 nm ± 10 nm) was integrated to induce photopolymerization of the printable matrix BioInk.

Image of the bioprinting device called 3DDiscovery used in the present study. (
Printing speed, frequency, as well as dispensed volume of each material are controlled by different parameters, such as dosing distance, valve opening time, and air pressure ( Table 1 ).
HMI Printing Parameters.

The bioprinting device is housed in a flow box permitting work under sterile conditions. The 3DDiscovery is capable of printing in standardized cell culture plates (Society for Biomolecular Screening standards) and cell culture inserts.
The HMI (human–machine interface) software of the 3DDiscovery bioprinter is the central tool to initialize and control the device as well as to define the printhead parameters ( Table 1 ).
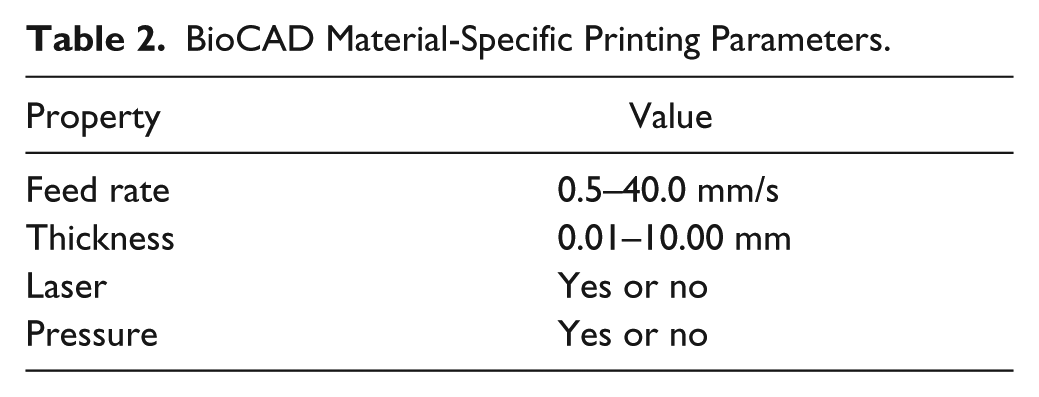
The BioCAD (version 1.0-040) program provided with the 3DDiscovery bioprinter allows the easy drawing of 3D patterns with defined dimensions for further fabrication of printed tissues. Each layer to be printed is clearly defined with respect to the shape and pattern, thickness, material, and printhead used. For each material, detailed parameters (feed rate, thickness, pressure, etc.) have to be defined ( Table 2 ). The printing sequence of the layers as well as the thickness of the designed model represent the last step of the BioCAD program before it is loaded in the HMI software for the 3D model realization.
BioCAD Material-Specific Printing Parameters.
Sterility Test
To assure sterile printing conditions, every component in contact with printed tissue was sterilized. The flow box of the bioprinter was cleaned with 75% ethanol and further sterilized by UV light for 30 min. The components to be assembled in the bioprinter, such as the disposable cartridges and the microvalves, were sterilized by autoclave and assembled in the pre-sterilized 3DDiscovery. Fibroblasts were expanded in monolayer cultures in HDFa medium I and detached by trypsin–EDTA before being printed or manually seeded in six well plates. The printing parameters used are summarized in Table 3 , and the dispensing process was repeated 10 times per well (about 50,000 cells/well in a total volume of 100 µl). Fibroblast viability was determined with the Cedex cell counter (Cedex Innovatis; Roche, Basel, Switzerland), and it was compared to the viability of manually seeded fibroblasts (positive control). Printed as well as manually seeded fibroblasts were cultured for 14 days at 37 °C and 5% CO2 in HDFa medium I (6 ml/well) without medium changes. Sterility was checked under the microscope.
Printing Parameters to Verify the Sterility of the Cell-Printing Process.
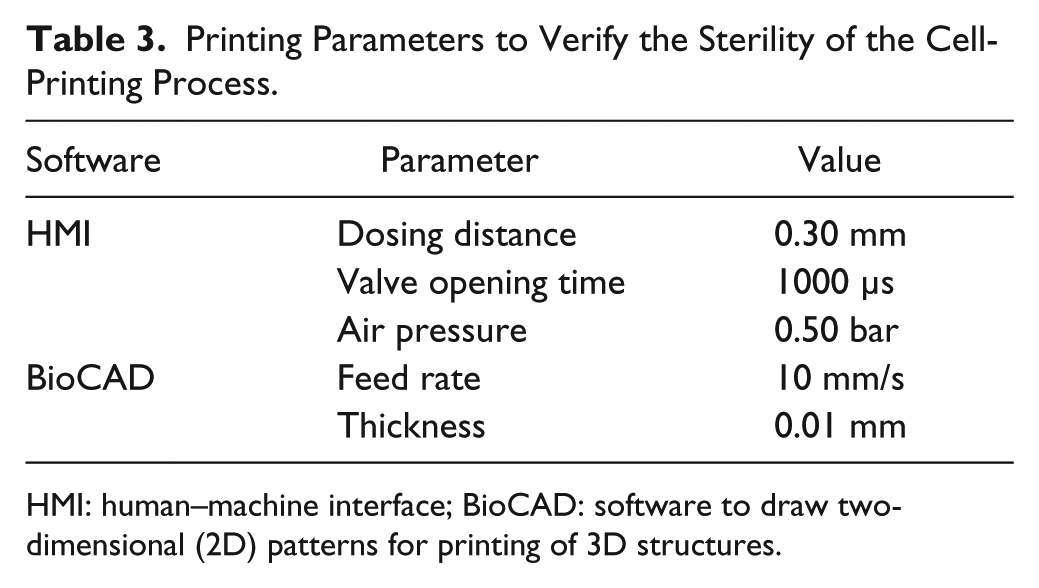
HMI: human–machine interface; BioCAD: software to draw two-dimensional (2D) patterns for printing of 3D structures.
BioInk
The printable ECM-like BioInk was developed and synthesized in collaboration with biologists and chemists at the Zurich University of Applied Sciences (ZHAW). To commercialize the BioInk, the production process was standardized, and the product is now being sold by regenHU Ltd. It is a ready-to-use, sterile, cell-compatible and -degradable solution. For fast polymerization, a cytocompatible photo-initiator is integrated that induces cross-linking of the chemically defined PEG-based BioInk on illumination at 365 nm ± 10 nm.
Fabrication of Dermal-Equivalent Models
Technical aspects and printing parameters
The BioInk solution was printed with the CF-300N printhead, which can dispense liquids with a viscosity of up to 1000
The fibroblasts were collected at a concentration of 9.0×106 cells/ml into 3 ml disposable syringes and were dispensed by jetting with the microvalve CF-300 nozzle diameter of 0.10 mm and stroke of 0.06 mm. The printing parameters defined in the HMI and BioCAD software are summarized in Table 4 .
Printing Parameters to Fabricate 3D Dermal Models.
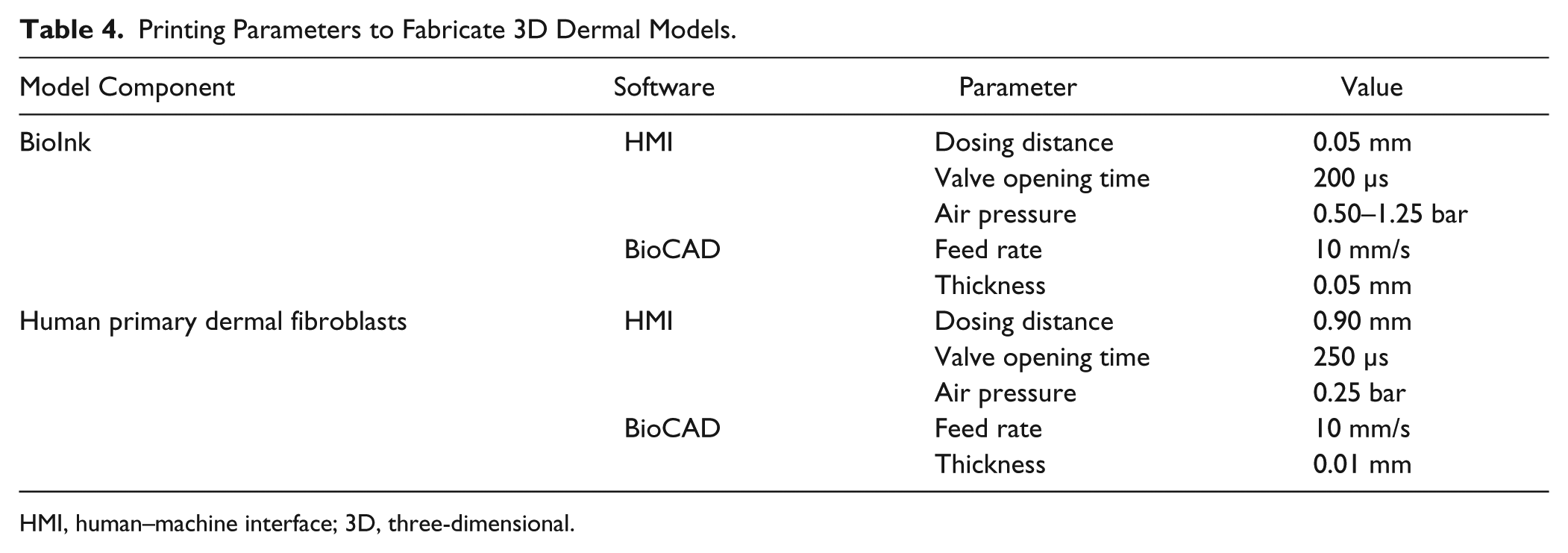
HMI, human–machine interface; 3D, three-dimensional.
Printing scheme and generation of 3D dermal-like models
The 3D dermis equivalent was constructed using the layer-by-layer approach. The 2D pattern of the structure was designed by the BioCAD software. The software allows the definition of printing direction and layer thickness.
Figure 2
shows the BioInk printing pattern as an angular spiral with external dimensions of 6×5.5 mm (
Fig. 2A
). BioInk layer thickness was set at 0.05 mm, and the first seven layers of BioInk were alternated with seven layers of printed cells (
Fig. 3
). Fibroblasts were printed in the same pattern as the BioInk with dimensions of 5×4.5 mm (
Fig. 2B
). The last printed cell layer was covered with an additional layer of BioInk to maintain the cells inside the model. For further experiments, including keratinocytes seeding, a cylindrical pattern consisting of seven BioInk layers with a radius of 2.7 mm was printed on top of the dermal model to provide a confined area for keratinocytes seeding. BioInk layer thickness was set at 0.05 mm (
Figs. 2C
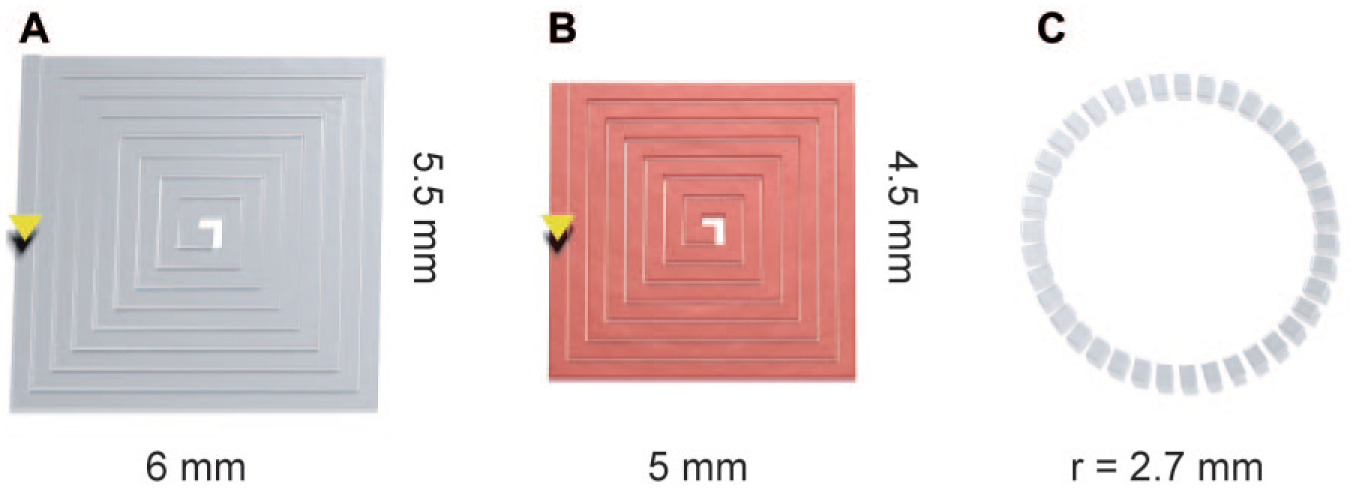
Scheme of the printing pattern in a layer-by-layer mode, as drawn by BioCAD software, for the generation of 3D dermal equivalents. The BioInk and the human primary fibroblasts were printed in an angular spiral pattern with dimensions of (

Schematic representation of printed 3D dermal models. (
The multilayered structures were printed directly on hydrophilic PTFE (polytetrafluoroethylene) cell culture inserts with 0.4 µm pore size (Millipore, Billerica, MA). The inserts containing the printed samples were transferred in six well plates and submerged in 6 ml of HDFa medium I supplemented with 1% penicillin–streptomycin (Sigma-Aldrich) and 0.1% fungizone antimycotic (Gibco) (HDFa medium II). The dermal models were cultured for up to 7 weeks at 37 °C and 5% CO2, and medium was changed every 2–3 days.
Cell Compatibility of Photopolymerization Process
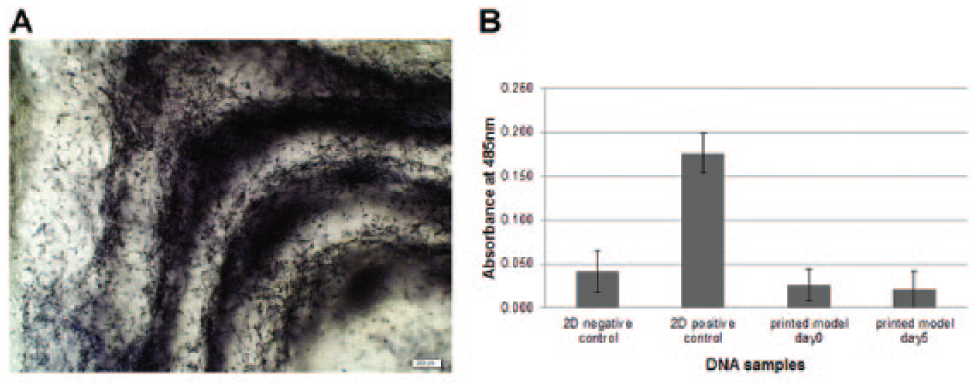
To verify the cell compatibility of the BioInk polymerization process, a cyclobutane pyrimidine dimer (CPD) enzyme-linked immunosorbent assay (ELISA) was performed. CPD formation is one of the predominant types of DNA damage caused by UV light. Dermis models were printed, and genomic DNA was isolated immediately after printing (day 0) and after 5 days of culture with the Wizard Genomic DNA Purification Kit (Promega, Fitchburg, WI) according to the manufacturer’s protocol. Each DNA sample (n = 3) was obtained from the processing of three different and pooled dermal models. Fibroblast DNA content was measured at 260 nm with a NanoDrop (Fisher Scientific, Hampton, NH). As blanks, 3D models were produced by replacing cell suspension with sterile water. In parallel, genomic DNA was isolated from fibroblasts cultured in 2D with and without UV exposure for 10 min at an intensity of 5 mW/cm2 (λ = 260 nm) to obtain positive and negative controls, respectively. The CPD detection was performed with the OxiSelect UV-Induced DNA Damage ELISA Kit (Cell Biolabs, San Diego, CA) following the manufacturer’s guidelines. The sample absorption was measured at 485 nm with a microplate reader (FLUOstar OPTIMA; BMG Labtech, Ortenberg, Germany). The blank absorption values were subtracted from the cell samples’ absorption values.
Viability Tests
MTT assay
Printed dermal models were cultured up to 7 weeks with medium changes every 2–3 days, and at specific time points (2 days, and 1, 2, 3, 4, 5, 6, and 7 weeks) the medium was replaced with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) 1 mg/ml in phosphate buffered saline (PBS). The samples were incubated for 3 h at 37 °C and 5% CO2. Only viable cells are able to reduce the MTT to insoluble formazan salts that stain cells blue, revealing their viability and distribution in the hydrogels. Samples were transferred in PBS for light microscopy analysis (Olympus 1X81 microscope equipped with the Olympus DP72 camera; Olympus, Tokyo, Japan).
PrestoBlue assay
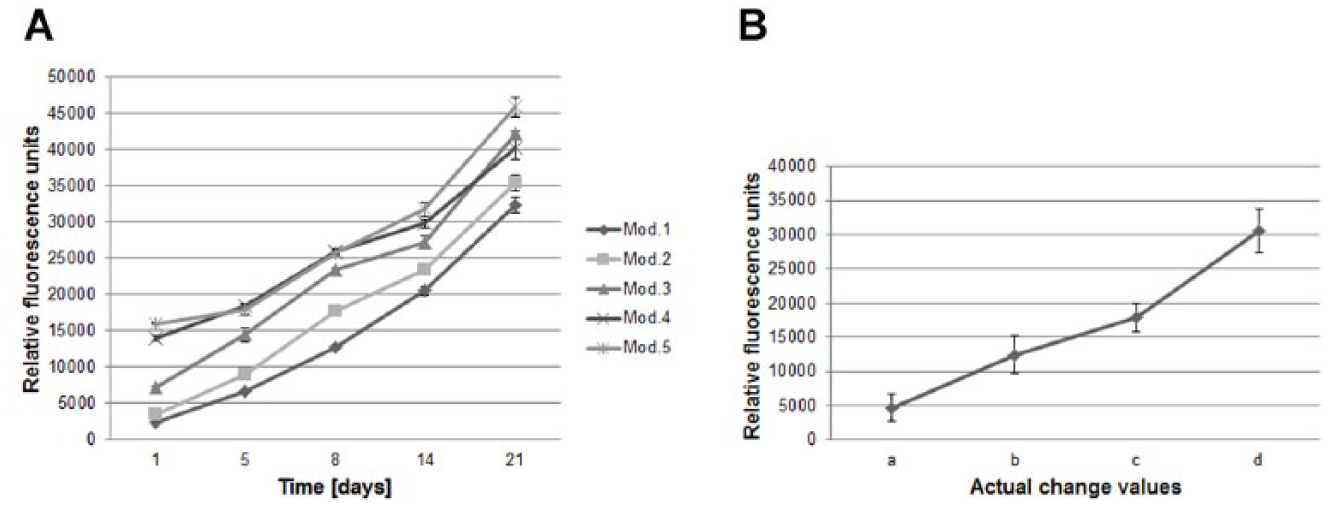
The PrestoBlue assay (Life Technologies, Carlsbad, CA) was performed with printed models harboring human primary fibroblasts and with “blank” printed models (cell suspension substituted with sterile water). Samples were analyzed at days 1, 5, 8, 14, and 21 after printing. The models were subcultured and measured again after each assay time point. The PrestoBlue reagent is a resazurin-based solution containing a nonfluorescent cell-permeable compound that turns fluorescent after being metabolized inside the cell. Only viable cells reduce the compound into the highly fluorescent resorufin. After each measurement, fresh medium was added, and the dermis models were subcultured while changing the medium every 2–3 days. For analysis, the HDFa culture medium II was replaced with PrestoBlue Cell Viability Reagent (Life Technologies) diluted 1:10 in culture medium. The samples were incubated for 2 h at 37 °C and 5% CO2, and media fluorescence was measured in the FLUOstar OPTIMA reader at λex 550 nm ± 10 nm and λem 590 nm ± 10 nm. The fluorescence readings [relative fluorescence values (RFVs)] of “blank” models were subtracted from the fluorescence values of dermal models. The test results were presented as a proliferation trend of each single model during the culture period ( Fig. 5A ) and as the average of the actual change value (ACV) of each printed model for the different time points ( Fig. 5B , where a = RFV samples day 5 – RFV samples day 1; b = RFV samples day 8 – RFV samples day 1; c = RFV samples day 14 – RFV samples day 1; and d = RFV samples day 21 – RFV samples day 1).
Collagen Type I ELISA
Human collagen type I was determined to evaluate the ECM production of fibroblasts in printed dermal models. Samples from culture supernatants were harvested for analysis at each medium change to calculate the amount of accumulation of produced collagen for up to 7 weeks. Collagen amount was correlated with metabolically active cells (PrestoBlue assay) populating the same printed dermis equivalents. Human collagen type I was determined using the MicroVue CICP EIA Kit (Quidel, San Diego, CA) according to the manufacturer’s instructions.
Full-Thickness Skin Model Production
Dermis equivalents were printed onto cell culture inserts and cultured in HDFa medium II for 7, 14, 21, and 42 days before human primary keratinocytes (HPEKs) were seeded on top to produce full-thickness skin models with dermal and epidermal parts. Before keratinocyte seeding, the HDFa medium II was completely removed from each well and from each cell culture insert. The gels were submerged in FTAL medium containing supplements (FTAL: Full-Thickness Air Lift; custom medium for full-thickness culture from CELLnTEC) for 2 h and were later exposed to air before seeding with keratinocytes. The dermis was air-dried under the bench for 15 min to improve further keratinocyte attachment. The HPEKs were detached by trypsin–EDTA, and 10 µl of a cell concentration of 107 cells/ml was seeded on top of each dermis model. The fibroblasts–keratinocytes–gels were incubated at 37 °C and 5% CO2 for 45 min to allow HPEK adhesion onto the gels, then they were submerged in culture for 4–5 days, changing the culture medium (FTAL medium with supplements) every 2–3 days. Cell culture inserts with the skin models were later transferred into petri dishes for further culture of the models up to 14 days at the air–liquid interface to induce keratinocyte differentiation into epidermis. Medium was changed three times per week.
Histological Analysis
Skin models harboring fibroblasts and keratinocytes were harvested after 14 days of culture at the air–liquid interface and were fixed in 10% neutral buffered formalin solution (Sigma-Aldrich) for 3 h at room temperature (RT) in the fume hood. Models were embedded in Tissue-Tek cryomedium (Sakura Finetek Europe, Alphen, the Netherlands) before freezing at −80 °C. Sections of 8 µm were prepared with Cryostat Microm HM 550 (Microm International, Waldorf, Germany) and were fixed for 10 min at RT in acetone–methanol (1:1) solution, precooled at −20 °C, before staining.
Hematoxylin and eosin (H&E) staining
Cryosections were incubated with CAT hematoxylin (Biocare Medical, Concord, CA) for 30 s, rinsed in tap water for 1 min, and incubated further in tap water for 5 min. The sections were transferred in 75% and 96% ethanol (2 min per solution) and stained with eosin-Y (J.T.Baker Chemicals, Avantor Performance Materials, Center Valley, PA) for 4 min. The samples were thoroughly rinsed with tap water and observed by light microscopy. As a reference tissue, a skin model purchased from MatTek (EFT312; MatTek, Ashland, MA) was sectioned and H&E stained.
Immunohistochemistry
To analyze keratinocyte distribution in the printed skin models, the cryosections were stained with monoclonal mouse antibody specific for cytokeratins 1, 5, 10, and 14 of the Moll catalog (Dako, Heverlee, Belgium). As positive controls, sections of epidermal models previously prepared according to protocols provided by CELLnTEC were used. Sections were incubated with antigen retrieval solution proteinase K (10 µg/ml in Tris–EDTA buffer pH 9.0; Sigma-Aldrich) for 10 min at 37 °C and rinsed three times with Tris-buffered saline (TBS). Sections were processed following the instructions of the Starr Trek Universal HRP Detection System (Biocare Medical). Briefly, sections were blocked for 10 min at RT with the Biocare Background Sniper solution and were then incubated for 1 h at RT with the above-described primary antibody (dilution 1:100 in TBS containing 1% bovine serum albumin). The sections were washed twice in TBS, incubated with Peroxidazed 1 (Biocare Medical) for 5 min at RT to quench endogenous peroxidase, and rinsed again twice with TBS before 20 min incubation at RT with Trekkie Universal Link (Biocare Medical). Sections were washed twice with TBS, incubated with TrekAvidin–horseradish peroxidase (HRP) solution for 10 min at RT, and rinsed twice with TBS before 5 min incubation with Betazoid DAB Chromogen (Biocare Medical) solution. Thereafter, sections were washed in tap water and counterstained with CAT Haematoxylin (Biocare Medical) for 30 s before another wash step in tap water and glycerol gelatin mounting (Sigma-Aldrich). The sections were analyzed by light microscopy.
Results
Cell Compatibility and Sterility of the Printing Process
A standardized procedure was established to assure high cell viability after the printing process. Human primary fibroblasts (HDFa) were automatically dispensed into tissue culture well plates according to printing parameters summarized in Table 3 , and viability was measured and compared to that of manually seeded cells. The printing parameters did not affect cell viability. Printed cells showed a high viability of 96.70% ± 5.45% versus 99.40% ± 0.42% of the positive control as determined with a Cedex cell counter. Moreover, culturing HDFa under standard (2D) conditions for 14 days demonstrated that printed cells presented the same morphology and ability to proliferate as manually seeded cells.
Sterility of the cell-printing process with the 3DDiscovery bioprinter was verified by culturing printed fibroblasts in HDFa medium I (proliferation medium without antibiotics) in well plates for 14 days without contamination.
BioInk Printing Features
The developed BioInk is provided as a ready-to-use sterile and printable solution in 3 ml cartridges. BioInk is a PEG-based matrix that is polymerized on illumination at 365 nm to obtain a chemically cross-linked hydrogel. The combination of high viscosity and fast photopolymerization allows high shape fidelity. In
Figure 3C
Characterization of Printed 3D Dermis Models
Having confirmed the viability of printed cells and the sterility of the printing process as well as the printability of the matrix, we further investigated the cytocompatibility of the BioInk. Human primary dermal fibroblasts were printed together with BioInk in alternating layers as shown in the scheme in
Figure 3B
to produce 3D dermal models (
Fig. 3C
Cell-compatible printing process. Human dermal fibroblasts were printed in a layer-by-layer mode with BioInk according to the printing scheme in
Figure 3A
. (
To verify the cell compatibility of the photopolymerization process using UV light at a wavelength of 365 ± 10 nm, a CPD ELISA test was performed. The use of UV light to polymerize the BioInk to produce tissue-like structures is essential, but cells are prone to DNA damage (mainly CPD formation) after UV exposure. Light exposition time and distance play fundamental roles for UV damage. Therefore, DNA integrity was assessed. Dermis models were printed, and genomic DNA of the encapsulated human dermal fibroblasts was isolated to analyze any CPD formation. Figure 4B shows the results obtained from analyzing the DNA isolated from printed dermal models directly after printing and after 5 days of incubation. As positive control for CPD formation, fibroblast cell suspension was exposed to UV light at 5 mW/cm2 and 260 nm wavelength. Cells in the printed models, independent of the time point analyzed, showed similar absorbance readings compared to the 2D negative control, in which fibroblasts were not exposed to UV light ( Fig. 4B ).
To investigate if the developed matrix could support not only fibroblast adhesion and spreading but also cell proliferation, 3D dermal models were printed and analyzed for up to 21 days with PrestoBlue Cell Viability Reagent. Five dermal models were printed, and proliferation was assessed in each model five times during the culture period. As negative control, BioInk structures were printed without cells. As shown in Figure 5A , the assay demonstrated that fibroblasts encapsulated in BioInk remained viable and proliferated for up to 3 weeks with increasing values of proliferation. The assay demonstrated a significant difference of the metabolically active cells and consequently of the cell number printed per model (see Fig. 5A ; fluorescence measurement at day 1 for each model). The fluorescence reading and consequently the cell number at day 1 in each printed model increased in the same sequence as the order in which the models were printed (day 1: model 1, 2480.30 ± 150.18 RFV; and model 5, 16027.90 ± 112.66 RFV). Although the initial cell number printed per model was not constant, the human primary fibroblasts were proliferating in the 3D printed BioInk structures throughout the time of the experiment ( Fig. 5A ). Interestingly, when averaging the ACVs of each printed model for the different time points, the models revealed a similar increase in proliferation as shown in Figure 5B . The models seeded with fibroblasts, unlike the blank structures (models without cells), remained stable for the entire culture period analyzed.
Viability and growth analysis of printed dermal models harboring human primary fibroblasts. (
In our study, printed dermal models were cultured for up to 7 weeks containing viable fibroblasts that were populating the entire printed scaffold as verified by MTT assay ( Fig. 6B ). In Figure 6A , an MTT-stained whole printed dermal model cultivated for 2 days is shown. The printing pattern was still visible, and the cells were spreading, confirming the results shown in Figure 4A . The fibroblasts inside the printed BioInk structure were not only proliferating but also producing their own ECM, as verified by the secretion in the culture medium of C-terminal of type I collagen peptide (CICP). Figure 6C shows the accumulated total protein amount increase during a culture time period of 7 weeks. The enhanced collagen I expression correlates with the increased cell proliferation inside the printed models as verified with PrestoBlue analysis. These findings confirm the biocompatibility of the PEG-based hydrogel and show its suitability for long-term 3D cultures.
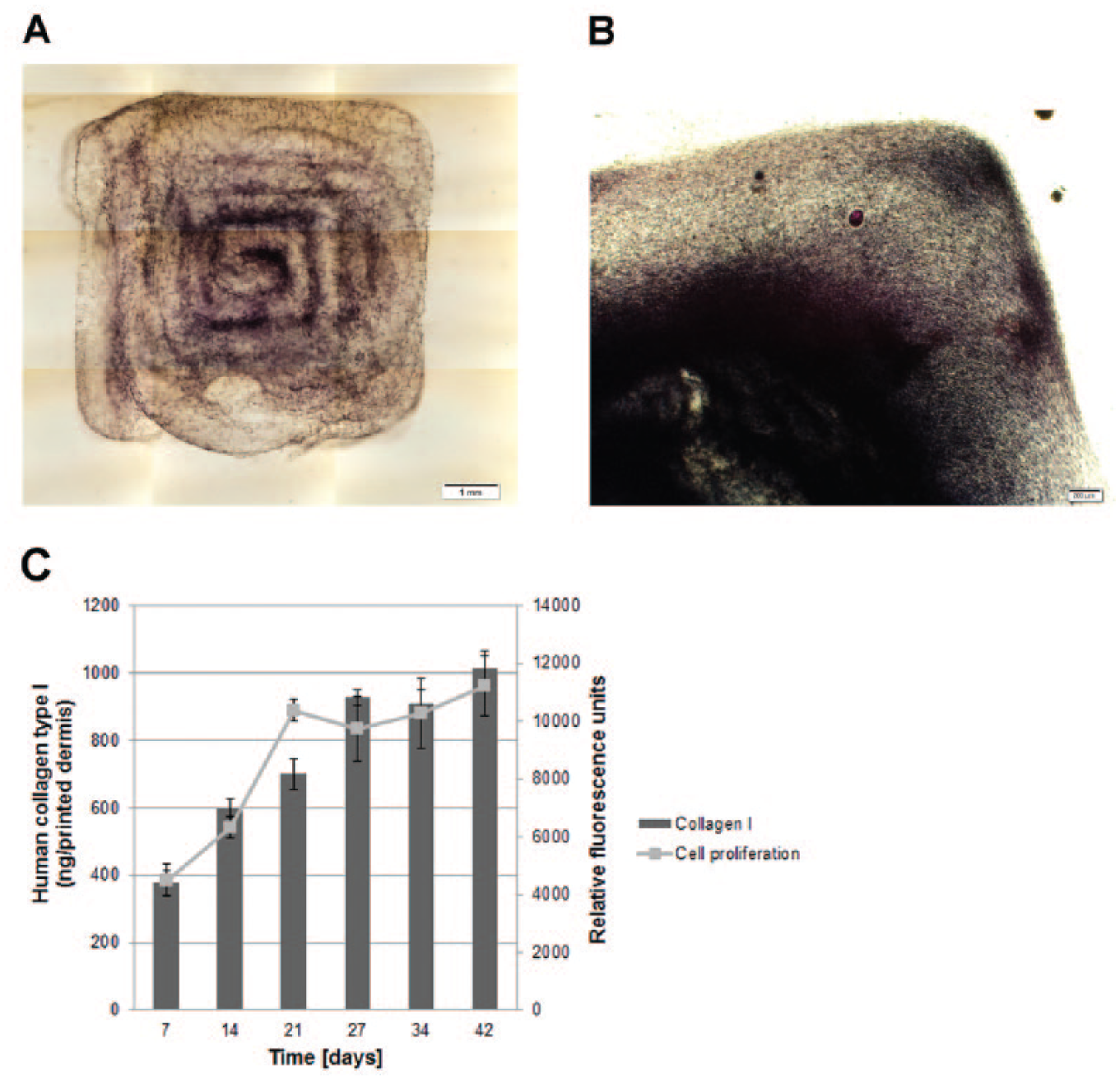
Long-term cultivation of printed dermis equivalents. (
Characterization of Printed 3D Skin Models
To develop a printed skin model harboring dermal and epidermal parts on top, we analyzed whether keratinocytes seeded onto the dermal models were able to adhere, proliferate, and eventually differentiate into a stratified epidermis. Dermal models were printed and cultured for 7, 14, 21, and 42 days before being seeded with human primary epidermal keratinocytes (HPEKs). Subsequently, the skin models were submerged in culture for 4–5 days prior to culturing at the air–liquid interface for 14 days to induce keratinocyte differentiation. During this incubation period, the cells in the models were fed solely from the bottom of the cell culture insert, leading to a reduction of model thickness in the first 5 days of air–liquid interface culture (data not shown). The models were fixed and prepared for cryosectioning before performing H&E staining on 8 µm slices to analyze their structure. Figure 7 shows H&E staining of skin models for which the dermal parts were cultivated for (B) 7, (C) 14, (D) 21, and (E and F) 42 days prior to keratinocyte seeding. The skin models were populated with many cells (blue nuclei). Eosin-stained structures (red) were distributed all over the skin models of dermal parts that were cultivated for 14, 21, and 42 days prior to keratinocyte seeding ( Fig. 7C–7E ; see arrows).
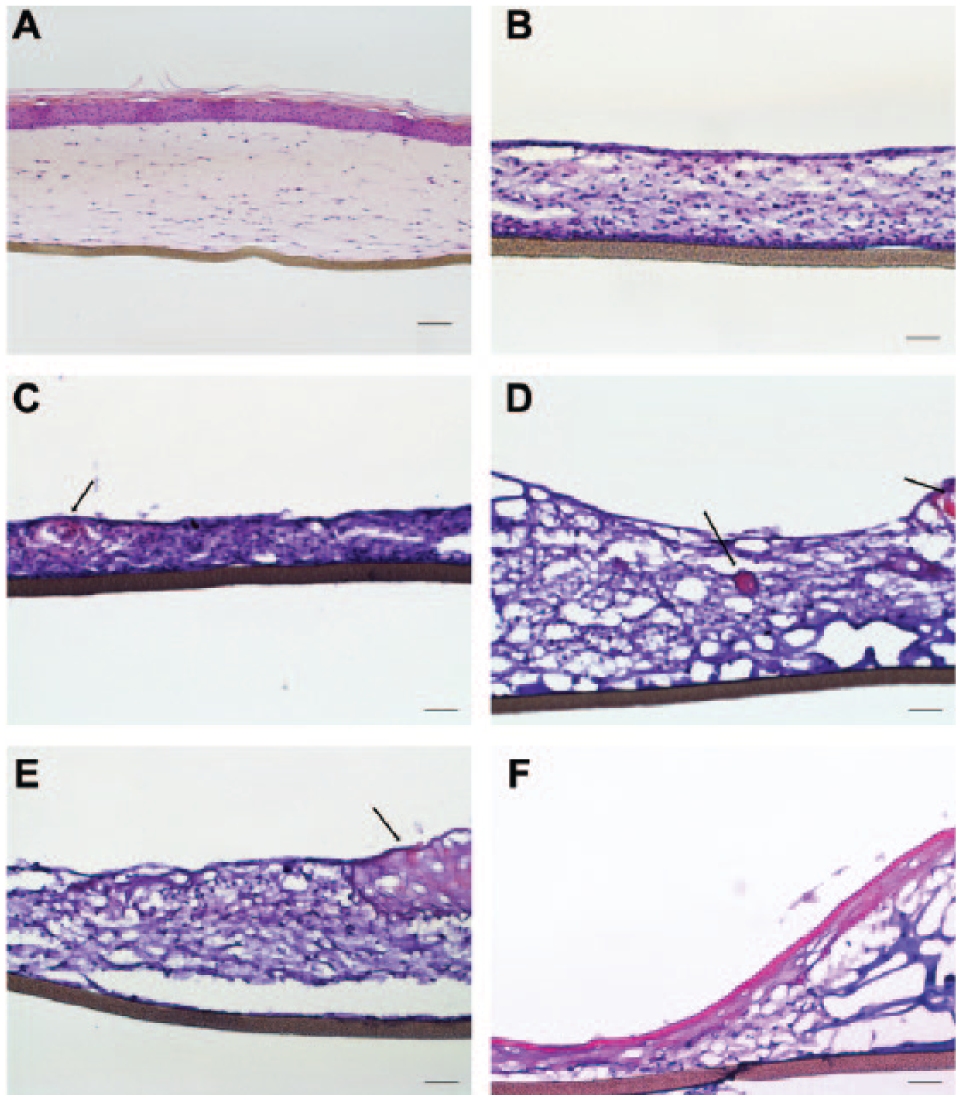
Hematoxylin and eosin (H&E) staining of printed skin models. (
A clear stratum corneum was visible in the skin models of dermal equivalents that were cultured for 6 weeks prior to keratinocyte seeding ( Fig. 7F ), whereas no epidermal layers developed in the earlier experimental conditions. For comparison, a commercially available skin model from MatTek (EFT312) was H&E stained, showing a well-differentiated stratified epidermis on top of the dermal part ( Fig. 7A ).
Cryosections of the printed skin models were further stained for cytokeratins solely expressed by keratinocytes to distinguish between dermal and epidermal layers. The antibody is specific for cytokeratins 1, 5, 10, and 14 of the Moll catalog. As a positive control, an epidermal model produced according to protocols published by CELLnTEC was used (
Fig. 8B
). In
Figure 8A
, raised epidermal models were stained without primary antibody to determine background levels of the staining (negative control). The antibody was specific for the epidermis visualized with the distinct brown staining (
Fig. 8B
). Cell nuclei were counterstained with hematoxylin to appear as blue points. We could demonstrate that the red (eosinophilic) structures previously detected by H&E staining inside the dermis (
Fig. 7
) were groups of keratinocytes (
Fig. 8D
and
8E
). In these conditions, the cells were not able to build an epidermis. In contrast, dermis cultured for longer times (e.g., 21 and 42 days) showed keratinocytes located on the surface, and a distinct epidermis, although not completely differentiated, was detectable (
Fig. 8 F–H
). These results showed a correlation between printed dermal culture incubation prior to keratinocyte seeding and epidermis formation. With time, keratinocyte migration into the printed dermis was decreased or even halted. The stratum corneum was clearly detectable on top of the model with the longest pre-incubation time of the dermis (
Fig. 8G
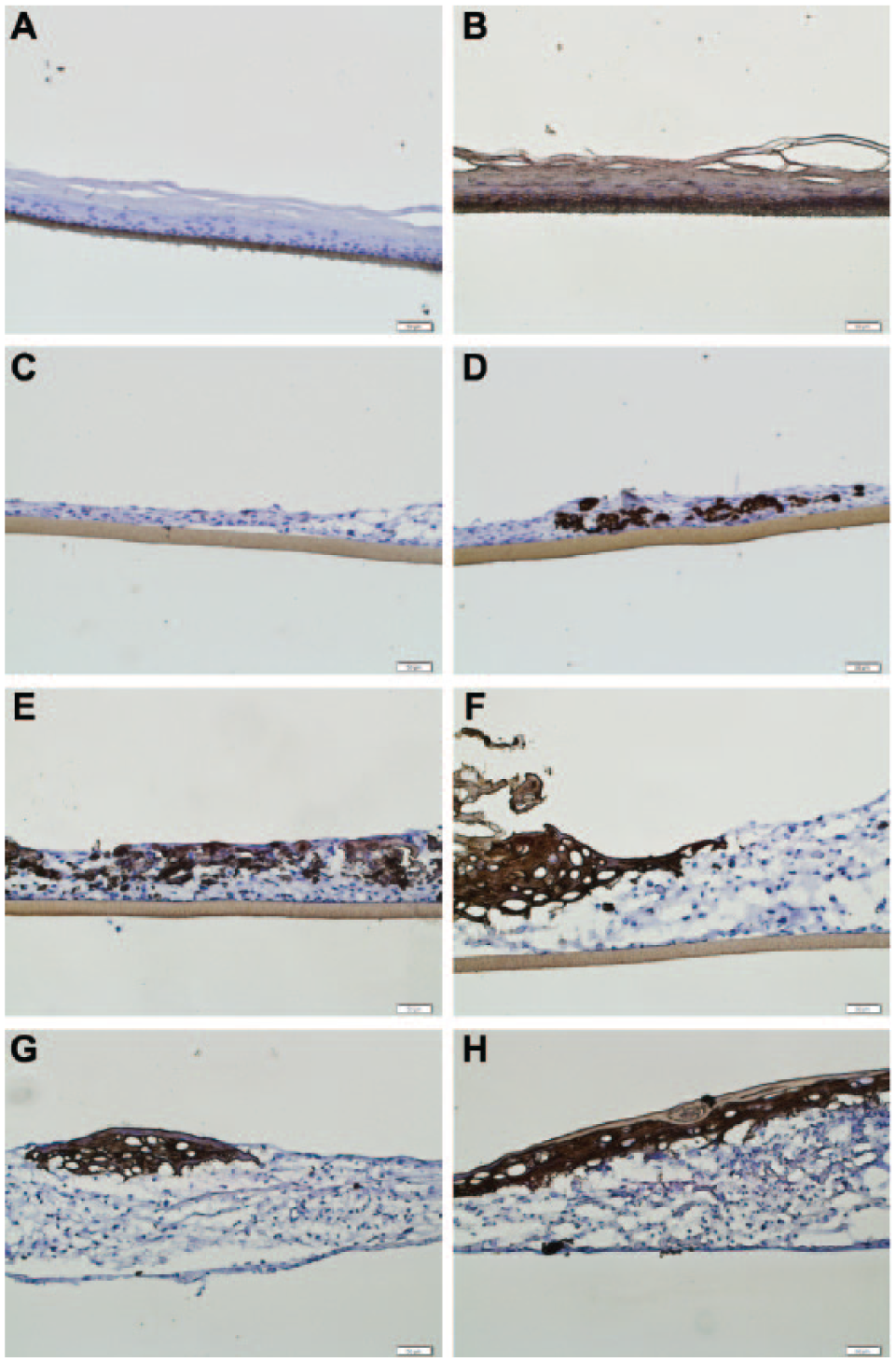
Immunohistological staining of printed skin models. Dermal equivalents were printed and cultured for (
Discussion
The bioprinting technology offers tremendous potential to produce reliable and complex organotypic tissue models for drug development and regenerative medicine.5–6,27 Different bioprinting technologies to deposit cells with high viability 28 as well as printable ECM-like hydrogels to support cell growth in 3D are available.9,27 However, at the moment, the bioprinting approach faces several shortcomings. Most printed tissue models are produced with in-house developed bioprinters, limiting the reproducibility and transferability of the achievements to other research groups or industries.29–30 Furthermore, commercialized bioinks are hardly available. Researchers active in this field must rely on biocompatible hydrogels, such as alginate, agarose, fibrinogen, collagen I, and matrigel, that have been proven valuable tools for culturing cells in 3D but are not optimized for printing applications. 10 An optimal bioink should provide not only a cell-compatible environment but also a high viscosity to print high-fidelity structures that are polymerized instantaneously after the printing process. 30 For this purpose, polymers, such as hyaluronic acid, dextran, or gelatin, are functionalized with acrylate–methacrylate groups to make them susceptible for photo-induced polymerization.29,31–33 Although these novel synthesized polymers show improved printing properties, the lack of a standardized production protocol hampers further industrial applications.
One of the first commercialized 3D bioprinters was provided by EnvisionTEC GmbH (Marl, Germany) and was used in many different studies.33–35 The instrument can process a variety of bioinks with light-, ionic-, or thermal-induced polymerization modes. To print under sterile conditions, the bioprinter is placed in the laminar flow bench.
The company Organovo Holdings (San Diego, CA) together with the company Invetech (Melbourne, Australia) developed a novel bioprinter capable of printing living tissues for medical research and therapeutic applications.17,36–37 The company provides a service to produce tissues from different cell types with their proprietary printing technology. The bioink consists of a “cellular paste” made of single or multiple cell types. This bioink is extruded in tissue-specific geometries together with sacrificial bioinks (e.g., agarose) as support material that is later removed to generate, for example, vascular structures with lumens. 17 After the printing process, the cells rearrange into a tissue-like structure. 17
In this work, we developed a holistic bioprinting approach, including (1) a commercialized bioprinter enclosed in a flow box for sterile tissue fabrication with a user-friendly software BioCAD, a photopolymerization unit (UV-LED; λ = 365 nm ± 10 nm), and cell-compatible printheads; and (2) a commercialized ready-to-use photopolymerizable BioInk. This all-in-one solution is supplemented with a standard operation procedure to generate printed soft tissue models. The inkjet-based printheads allow printing solutions in viscosities up to 1000 mPa . s. In a proof-of-concept study, a skin-like tissue model was printed as a soft tissue reference.
To produce skin equivalents, we used in our study human primary cells that, unlike cell lines, have the potential to fully differentiate into a stratified epidermis. 38 However, primary cells are as well known to be more sensitive than cell lines. 39 In standard skin equivalent production, fibroblasts are embedded in collagen I. 40 Although collagen I is also used in other skin bioprinting approaches,24,26 we developed the BioInk as ECM-like matrix with improved printing properties and without batch-to-batch variations. 41 A cell suspension of human dermal fibroblasts was printed in alternating layers with BioInk in a predefined pattern as shown in Figure 3 to represent the dermal compartment. The fibroblasts in the printed dermis were viable and cultured up to 7 weeks, thereby populating the entire printed structure. Despite the high sensitivity of primary cells, the established printing conditions as well as the developed BioInk turned out to be cell-friendly. In addition, the use of UV light to induce BioInk polymerization did not affect the DNA integrity of the fibroblasts as verified with analysis of CPD generation. UVA light (320–400 nm) is known to induce CPDs as well as double-strand breaks in skin tissue.42–44 Moreover, fibroblasts not only were able to adhere, spread, and remain viable inside the constructs but also were proliferating as shown with viability tests for up to 42 days. When comparing printed acellular with printed fibroblast-containing models, the cell-containing tissues were long-term stable, which can be attributed to the cells’ collagen I expression leading to strengthening and remodeling of the printed structure. With respect to drug development, the feasibility for long-term cultures of organotypic models is an important aspect allowing repeated drug exposure to analyze chronic drug toxicity. 45
Fundamental for epidermis formation is the initial adherence of keratinocytes on the surface of the dermis to build a monolayer. To address this issue, Lee and coworkers 24 were printing keratinocytes (HaCaT) in collagen I hydrogels to immobilize the cells on top of a dermal part. Using LaBP, Koch et al. 25 transferred the keratinocytes previously embedded in collagen I hydrogels onto printed dermis. However, it is not reported whether the embedding of keratinocytes into a matrix interferes with epidermis formation. In this study, a cylindrical structure, as shown in Figure 3 , was printed with BioInk on top of the printed dermis to provide a container for keratinocytes seeding to prevent cell loss. Keratinocytes strongly depend on secreted factors and ECM produced from fibroblasts to properly differentiate into a stratified epidermis.46–48 Thus, printed dermal models were cultured for up to 7 weeks and were seeded with human dermal keratinocytes at different time points to determine the optimal seeding procedure. In the printed dermal models, ECM expression increased throughout culture time as verified with collagen type I expression. As expected, this finding correlated with improved cell attachment and epidermis formation at later keratinocyte-seeding time points. In short-term cultivated dermal models, the keratinocytes tended to migrate into the dermis.
Despite these promising results in standardized bioprinted organotypic tissue production, further optimization is necessary to transfer the process into industrial applications. In our study, a fully stratified epidermis as obtained with standard skin equivalent protocols using collagen I 40 was not achieved. Furthermore, the number of printed cells should be constant in each model to obtain standardized dermis equivalents. In our approach, fibroblast number increase during culture time was constant among the dermal models but not initial cell numbers. This problem is referred to as “cell sedimentation” during the printing process due to the static maintenance of cells in aqueous low-viscosity solutions. We are currently evaluating different stirring systems and velocities in our group to obtain a homogeneous and viable cell suspension during the entire printing process. The sedimentation effect has also been described by Parsa et al. 49 They introduced gentle agitation and surfactant addition into the medium during printing to maintain the cells suspended and improve droplet integrity. Although a homogenous cell suspension was achieved, cell aggregation hindered extended printing times. Chahal et al. 50 were increasing the viscosity of the cell suspension with Ficoll PM400 to obtain neutral buoyancy, thereby reducing cell sedimentation in the printing cartridge. In general, increasing viscosity reduces cell sedimentation, thereby enhancing shear forces, which might have deleterious effects on the cells. 51
In our study, keratinocytes were seeded manually on top of the dermal constructs to precisely deposit the cells. The reason for manual cell pipetting was the lack of an optical system to, for example, exactly locate previously printed structures, allowing the manipulation of printed constructs after a certain culture time. This feature should be integrated in next-generation bioprinters and is under investigation in our lab. Efforts are also put into the development of cell-type-specific bioinks that provide all the necessary cues for proper cell functioning. 9 This results in extensive characterization and standardization of novel bioinks. 9 The here-developed BioInk showed compatibility with several cell lines and primary cells (data not shown) exhibiting universal properties.
In summary, although in bioprinting several technical and biological issues still need to be tackled, the work presented here will help in promoting this technology to find its way into routine industrial applications.
Conclusion
We have presented, for the first time, a bioprinting all-in-one solution to produce a skin-like reference soft tissue model harboring human primary fibroblasts and keratinocytes. The established printing process and the developed BioInk are cell compatible and permit the generation of long-term culture models. Further optimization of our system can potentially facilitate the complete standardization of 3D tissue model generation. Future advancements of this technology will rely on standardization of bioprinting devices and customized bioinks to promote proper cell function. Especially in the cosmetic industry, reliable in vitro skin models are urgently needed for the testing of cosmetic ingredients as demanded by law in Europe. Also in drug development, the production of 3D tissue equivalents with bioprinting exhibits a huge potential. Especially, long-term culture of in vitro tissue models is of interest to monitor the effects of multiple drug administration. Furthermore, regenerative medicine will benefit from the bioprinting technology because it offers the possibility to reproduce tissue and organ complexity in vitro. The implementation of a vasculature system into the printed tissue will allow the production of larger tissues.
Footnotes
Acknowledgements
We gratefully thank the innovation promotion agency (CTI, Switzerland, Project Nos. 12148.2 and 14331.1) for financial support of the project and technical guidance. We thank Prof. Dr. Rainer Riedl (ZHAW, ICBC, Center for Organic and Medicinal Chemistry) for chemical synthesis support.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors wrote this article as part of their employment with the Institute of Chemistry and Biological Chemistry (ICBC), Zurich University of Applied Sciences (ZHAW), Waedenswil, Switzerland.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We gratefully thank the innovation promotion agency (CTI, Switzerland, Project Nos. 12148.2 and 14331.1) for financial support of the project and technical guidance.