
Abstract
Mitochondria harbor the oxidative phosphorylation (OXPHOS) system, which under aerobic conditions produces the bulk of cellular adenosine triphosphate (ATP). The mitochondrial genome encodes key components of the OXPHOS system, and it is transcribed by the mitochondrial RNA polymerase, POLRMT. The levels of mitochondrial transcription correlate with the respiratory activity of the cell. Therefore, compounds that can increase or decrease mitochondrial gene transcription may be useful for fine-tuning metabolism and could be used to treat metabolic diseases or certain forms of cancer. We here report the establishment of a novel high-throughput assay technology that has allowed us to screen a library of 430,000 diverse compounds for effects on mitochondrial transcription in vitro. Following secondary screens facilitated by the same assay principle, we identified 55 compounds that efficiently and selectively inhibit mitochondrial transcription and that are active also in cell culture. Our method is easily adaptable to other RNA or DNA polymerases and varying spectroscopic detection technologies.
Keywords
Introduction
As with many other classes of enzymes, classical approaches to measure the activity of DNA and RNA polymerases rely on the detection of product accumulation. This is usually accomplished either by incorporation of radioisotope-labeled nucleotides for direct detection of DNA/RNA species or via amplification of said DNA/RNA and detection of the resulting oligonucleotide copies by the use of quantitative real-time PCR (qRT-PCR). Both of these methods have their strengths in high specificity and the attainable sensitivity, but they also have clear disadvantages in that they are heterogeneous, rely on noxious and costly reagents, or are amenable only to batch-wise processing of samples. All of these factors are obstructive for the smooth conduct of a fully automated high-throughput screen and have restricted the use of these methods, with few exemptions, to applications with lower sample numbers.
Measurement of DNA and RNA polymerase activities can be used for the identification of new inhibitors of bacterial or viral growth. This type of assay, however, also may be used to monitor for adverse effects on cellular polymerases. Before the recent success of sofosbuvir, a number of other, promising nucleoside inhibitors (NIs) directed toward the hepatitis C RNA polymerase NS5B were stopped during clinical development due to serious adverse side effects. 1 Valopicitabine, a prodrug of 2’-C-methyl cytidine (2’CMeC), was halted due to dose-dependent gastrointestinal toxicity,2–3 whereas VX-135 (2’Me/OH-O-UTP) was halted during phase II development due to hepatotoxicity. 1 In fact, many NIs developed to treat viral infections have turned out to be substrates for the human mitochondrial RNA polymerase (POLRMT), and the efficacy of incorporation in vitro correlates with effects on mitochondrial transcription in vivo. 4 Antiviral NIs are often used for long periods of treatment (often more than 12 weeks), and compounds that are poor substrates for POLRMT in vitro are less likely to show mitochondrial toxicity in vivo. Based on these observations, a screening paradigm to predict the mitochondrial toxicity of NIs has been proposed, which includes measurement of POLRMT activity in vitro coupled with monitoring of mitochondrial protein production and cellular respiration. 4
We wanted to combine the specificity and sensitivity of more classical means of measurement with the advantages of a homogeneous detection procedure. To this end, we set out to develop a novel assay system that would be amenable to high-throughput screening (HTS) and applicable to the mitochondrial transcription machinery. We reasoned that the flexibility and swiftness of oligonucleotide synthesis and coupling should greatly facilitate the method development process and in general ensure adaptability and cost-effectiveness of the resulting assay. Although the described method is applicable to a wide variety of RNA and DNA polymerases, the initial aim of our method development was to measure the activity of the human POLRMT. POLRMT is a single-subunit RNA polymerase that is similar in sequence and structure to the bacteriophage T7 RNA polymerase. 5 POLRMT cannot initiate promoter-dependent transcription on its own but requires two additional transcription factors, transcription factor A (TFAM) and transcription factor B2 (TFB2M). These factors are needed for POLRMT recruitment and the initial melting of DNA in the double-stranded promoter region. Based on the protocol for in vitro reconstitution of the minimal mitochondrial transcription machinery,6–7 we first chose the mitochondrial “light-strand” promoter (LSP) as a template sequence for the transcription reaction.
In the assay reported here, we detect nascent RNA by functionalized DNA oligonucleotides in a sequence-specific fashion. These oligonucleotide probes selectively bind to two consecutive sequence stretches of the RNA, such that the respective functionalized 3′ and 5′ termini of the oligonucleotides are brought in close proximity. By choosing dyes with suitable absorption and emission spectra as functional groups of the respective probes, a fluorescence donor–acceptor couple can be constructed. Consequently, a Förster resonance energy transfer (FRET) signal is generated on excitation of the donor dye, only in case both probes are bound to the RNA. The specific design of the described method facilitates the generation of a time-resolved FRET signal (TR-FRET), which reflects the time-dependent accumulation of the target transcript and is thus a direct measure of the activity of POLRMT. The detection method in particular proved to be very robust against diverse reaction conditions and was applicable to a wide pH range and wide variations in salt and DMSO concentrations.
To validate this novel method, we assessed how the mitochondrial transcription reaction can be modulated, depending on reaction temperature, substrate, template DNA and POLRMT concentrations, or the influence of small molecule binders. We also used our method to monitor the effects of a number of NIs.
Materials and Methods
Recombinant Proteins
Human POLRMT, TFAM, and TFB2M were expressed and purified as described in Ref. 7 .
In Vitro Mitochondrial Transcription Assay
A DNA fragment corresponding to positions 1–477 of the human mitochondrial DNA (mtDNA) cloned into a pUC18 plasmid was linearized with Bam HI and used as a template for in vitro transcription to produce a run-off product of ~400 nt. 6 All transcription reaction volumes were 25 µL and contained 10 mM Tris-HCl pH 8.0, 10 mM MgCl2, 68 mM NaCl, 100 µg/mL bovine serum albumin (BSA), 1 mM DTT, 4 U RNase inhibitor Murine (New England Biolabs, Ipswich, MA), 100 fmol of linearized human LSP template, 400 fmol POLRMT, 600 fmol TFB2M, and 5 pmol TFAM. When we used radioactive uridine triphosphate (UTP), the reactions also contained 400 µM ATP, 150 µM cytidine triphosphate (CTP), 150 µM GTP, 10 µM UTP, and 0.02 µM α-32P UTP (3000 Ci/mmol). When we used radioactive guanosine triphosphate (GTP), the reactions contained 400 µM ATP, 150 µM CTP, 10 µM GTP, 200 µM UTP, and 0.02 µM α-32P GTP (3000 Ci/mmol). The reactions were stopped after 30 min at 32 °C by the addition of stop buffer (10 mM Tris-HCl pH 8.0, 0.2 M NaCl, 1 mM EDTA, 0.1mg/ml glycogen, and 100 µg/mL proteinase K) followed by incubation at 42 °C for 45 min. The transcripts were purified with ethanol precipitation, and the pellets were dissolved in 20 µL gel loading buffer (98% formamide, 10 mM EDTA, 0.025% xylene cyanolFF, and 0.025% bromophenol blue) and heated at 95 °C for 5 min. The samples were analyzed on a 4% denaturing polyacrylamide gel (1× Tris–borate–EDTA buffer with 7 M urea) followed by exposure on film.
Homogeneous TR-FRET Assay for HTS and Activity Determination
The protocol described here was applied for screening and activity determination in a low-volume 384-well microtiter plate with a nonbinding surface. For high-throughput application in the 1536-well microtiter plate format, volumes of the reagent mixes were adjusted, maintaining the volumetric ratio. Proteins POLRMT, TFAM and TFB2M were diluted from their stocks to working concentrations of 150 nM, 1.8 µM, and 330 nM, respectively, in a dilution buffer containing 100 mM Tris-HCl pH 8.0, 200 mM NaCl, 10 % (v/v) glycerole, 2 mM glutathione (GSH), 0.5 mM EDTA, and 0.1 mg/mL BSA. Protein dilutions and template DNA, comprising a pUC18 plasmid encoding the mitochondrial light strand promoter, restriction linearized proximal to the promoter 3′-end (pUC-LSP), were mixed at the twofold final assay concentration in a reaction buffer, containing 10 mM Tris-HCl pH 7.5, 10 mM MgCl2, 40 mM NaCl, 2 mM GSH, 0.01% (w/v) Tween-20, and 0.1 mg/mL BSA.
Five microliters of this mix were dispensed, depending on the chosen microtiter plate format, using multichannel pipettes or a Multidrop dispenser (Thermo Fisher Scientific, Waltham MA) into the wells of a microtiter plate and incubated at room temperature (RT) for 10 min. Chemical compounds under scrutiny in the assay were applied using a contact-free acoustic droplet-dispensing Echo520 (Labcyte Inc., Sunnyvale CA) from 10 mM compound stocks in 100% DMSO, to a final concentration of 10 µM or in serial dilution series of the required concentration range. Equal amounts of DMSO without any compound were added to positive control samples, followed by an incubation step at RT for 10 min.
The enzymatic reaction was started by the addition of 5 µL of a mix of deoxyribonucleotide triphosphates (dNTPs) in reaction buffer to a final concentration of 500 µM each. No nucleotide mix was added to negative control samples. The content of the wells was mixed using a VarioTeleshaker (Thermo Fisher Scientific) at 1500 rpm for 45 sec, after which the microtiter plate was centrifuged at 500 ×g for 1 min. The samples were incubated for 2 h at RT with humidity control to avoid evaporation. The detection reagents were prepared in a buffer that was composed so that the enzymatic reaction was terminated due to chelating of Mg-ions and increased ionic strength; it contained 50 mM Tris-HCl pH 7.5, 700 mM NaCl, 20 mM EDTA, and 0.01% (w/v) Tween-20. Eu-cryptate-coupled streptavidin had to be preincubated with a 100-fold molar excess of a random sequence oligonucleotide for 10 min at RT in the dark to block unspecific binding of single-stranded RNA to the protein. Subsequently, the blocked streptavidin(–Eu) was mixed with the DNA probes on ice and kept away from light until use.
At the end of the enzymatic reaction time, 10 µl of detection reagent mix was added, such that the final concentration of fluorescent-donor probe (bio-5′-AACACATCTCT(-bio)GCCAAACCCCA-bio-3′), fluorescent-acceptor probe (ATTO647N-5′-ACAAAGAACCCTAACACCAG-3′), and streptavidin(–Eu) in each assay well was 1 nM, 3 nM, and 1 nM, respectively. Assay plates were again mixed and centrifuged as above and stored at RT, protected from light for at least 2 h or until binding of the DNA probes to RNA product and binding of streptavidin(–Eu) to the biotinylated DNA probe led to the development of the maximal FRET signal. The generated signal was measured with an EnVision plate reader, including a TRF light unit (Perkin Elmer, Waltham, MA), using excitation at 320 nm, an integration time of 200 µs, and a delay time of 100 µs, prior to detection at 620 nm and 665 nm. The ratio of donor to acceptor fluorescence was used to assess the specific FRET signal as a measure of the generated product content (i.e., enzymatic activity).
A Generic Homogeneous TR-FRET Assay to Assess the Activity of RNA Polymerases without Ancillary Factors
The protocol was adapted from the method described above, such that instead of a linearized plasmid comprising a promoter sequence, a “tailed” DNA oligomer was used as a template for in vitro transcription. The template is a 114 bp long double-stranded DNA (dsDNA) construct with a 14 nt single-stranded 3′-overhang, allowing for binding of RNA polymerases. The tailed template was generated by primer annealing of the following two oligonucleotides: 5′-GGC GGG AGA AGA ATT TGA AAT CTG GTT AGG
Quantitative RT-PCR to Assess Cellular Activity
qRT-PCR, based on the TaqMan (Thermo Fisher Scientific, Waltham MA) technology, was carried out essentially as described in Ref. 8 . HeLa cells were plated 1 day before compound treatment in RPMI medium supplemented with 10% fetal calf serum and 2 mM L-glutamine. Cells were incubated with dilution series of compounds or vehicle (DMSO) for 4 h, prior to harvest and extraction of the RNA using the RNeasy Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. RNA concentrations were measured spectroscopically using a NanoDrop-2000 (Thermo Fisher Scientific), and they were normalized prior to complementary DNA (cDNA) synthesis using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qRT-PCR was carried out using the TaqMan Fast Advance Master Mix (Thermo Fisher Scientific) on a 7500 Fast Real-Time PCR machine (Applied Biosystems, Foster City, CA).
For these measurements, three genes were used to compare the effects of the scrutinized compounds in relation to their concentration. The POLRMT gene was used to detect potential influences on nuclear transcription. Mitochondrial transcription in vitro was monitored by measurements 7S RNA. The TBP (TATA-box binding protein) gene was used as the control (housekeeping gene) during qRT-PCR. The short-lived mitochondrial 7S RNA, which is not posttranscriptionally stabilized, allowed us to monitor rapid changes in mitochondrial transcription activity following compound addition. Biological triplicates were analyzed using the comparative CT method (ΔΔCT) method 9 and reported as Rq% values (Rq = relative quantification = 2-ΔΔCT).
Results
To monitor mitochondrial transcription, we used a dsDNA fragment containing the mitochondrial LSP. Run-off transcription from the promoter produced a 407-nt RNA fragment. For detection of the nascent RNA, we used two functionalized DNA oligonucleotides (20 and 22 nt), which bound sequence-specifically to two consecutive regions of the transcript. To detect binding of the probes and hence determine RNA levels, we introduced a fluorescence donor–acceptor pair on the two oligonucleotides. Once bound to RNA, the respective functionalized 3′ and 5′ termini of the oligonucleotides are separated by only three nucleotides (~1–3 nm), which allows for the generation of a time-resolved FRET signal ( Figure 1A ). One oligonucleotide was labeled with biotinylated thymidine bases, whereas the other oligonucleotide was labeled with the fluorescent probe ATTO647N (ATTO-TEC, Siegen, Germany). Following binding of the DNA probes, Europium-cryptate coupled streptavidin (Cisbio, Codolet, France) was added and bound to biotin. Eu-cryptate was next excited at 320 nm to emit a fluorescence signal at 620 nm after 1–2 msec. Only when the ATTO647N label is found in close proximity (< Förster radius) to the Eu-cryptate can radiation free energy transfer occur between the fluorescence donor (Eu) and the acceptor (ATTO647N) and a time-resolved signal at 665 nm is generated. Although the direct coupling of the oligonucleotide probes with, for example, Eu-cryptate and suitable fluorescence acceptors also led to the successful establishment of a FRET pair, we observed the highest signal intensities and best overall assay performance when using a combination with functionalized streptavidin. The latter, however, had to be preincubated with unbiotinylated random oligonucleotides to saturate interfaces for unspecific binding of RNA and DNA on the protein surface.
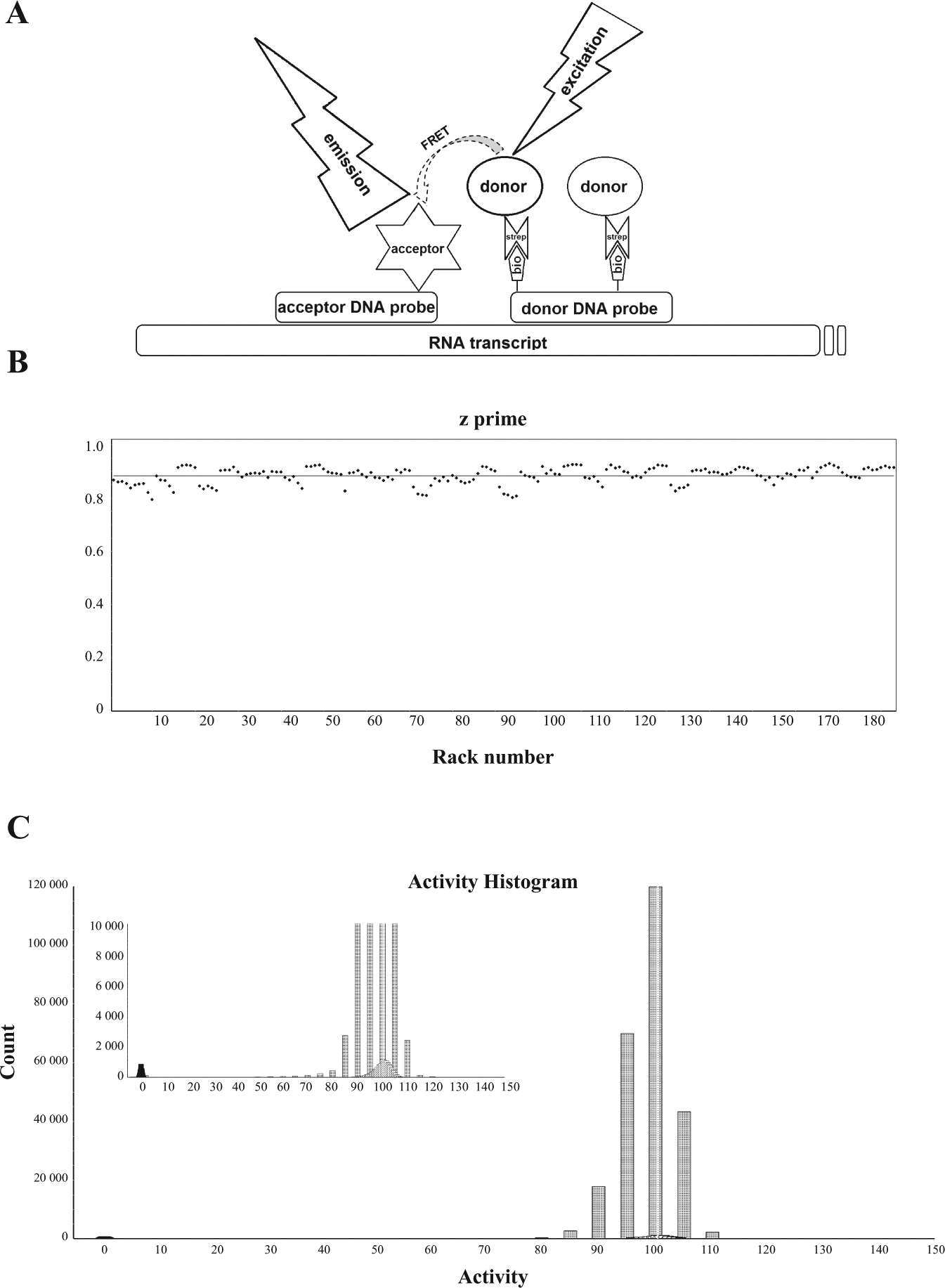
A high-throughput assay for mitochondrial transcription. (
We first applied the developed assay method in a true high-throughput mode, screening a diverse library of small chemical compounds to identify inhibitors of the human mitochondrial transcription machinery. About 430,000 substances were tested in a 1536-well microtiter plate format using contact-free acoustic compound transfer as well as predispensed compound dry films in assay-ready microtiter plates. We observed excellent assay performance (signal-to-background ratio >10 and signal-to-noise ratio ~50) and robustness, as expressed in a mean z’ value of >0.87, shown for a sublibrary of 250,000 compounds ( Figure 1B ).
The overall activity distribution of the screened assay wells represents an almost ideal, “Gaussian” curve ( Figure 1C ), with only a slight shift of the activity distribution toward lower values. Consequently, the threshold for hit selection was set stringently to 30% residual activity, relative to the control (100% DMSO). Thus, after filtering out substances that displayed autofluorescence or fluorescence-quenching properties in the excitation or emission wavelength of the assay, 580 primary hits were selected for further analysis, corresponding to a hit rate of 0.13%. Primary hit compounds were further scrutinized using a triage of different validation assays. Thus, dose-dependent activity was assessed in the described assay format, using a serial compound dilution series between 30 µM and 14 nM. Substances displaying IC50 values lower than 10 µM were subjected to structural clustering and filtering based on established chemo-informatics criteria.10–13
If provided with a dsDNA template containing a single-stranded 3′ tail, POLRMT can initiate transcription independently of the other components of the mitochondrial transcription machinery (i.e., TFAM and TFB2M). 6 Tailed templates are therefore ideal to monitor the direct effects on POLRMT activity independent of promoter recognition. We therefore adapted our assay method and used a ~100 bp dsDNA construct with a 14 nt single-stranded overhang as template. With this assay, we investigated a set of 280 selected substances that had been identified as inhibitors of mitochondrial transcription in our initial HTS. Although the efficiency of the transcription reaction on the tailed template and, consequently, the resulting signal intensities were somewhat reduced (signal-to-background ratio ~2.5), the observed assay robustness was very good, allowing us to identify 180 direct inhibitors of POLRMT in a swift high-throughput fashion.
A tailed template can be used as substrate for many RNA polymerases. We thus reasoned that our adapted assay should be generic enough to also allow assessment of the selectivity of the identified inhibitors against other polymerases. Indeed, we established two further variations of the adapted assay protocol with minimal effort and were, thus, able to discriminate selective POLRMT inhibitors from substances that also inhibited RNA polymerases of E. coli and bacteriophage T7 in a dose-dependent fashion (data not shown). Of the 180 substances scrutinized, only 50 different compounds yielded a meaningful dose response when tested against bacterial and viral RNA polymerase at a maximum concentration of 50 µM. Two known inhibitors of bacterial RNA polymerase (aureothricin and thiolutin) and the virostatic amantadine, which were used as reference substances in the respective assays, showed no effect when tested in dose–response measurements against POLRMT (data not shown).
For an independent validation of the identified inhibitors, substances with proven selectivity were further scrutinized in an orthogonal biochemical assay system, which did not rely on hybridization for detection of transcription products. To this end, radiolabeled nucleotide triphosphates were incorporated in an in vitro mitochondrial transcription reaction, which enabled direct detection of nascent RNA.
6
Compounds were tested for their dose-dependent inhibition of the transcription reaction. The observed inhibition was in excellent agreement with data obtained in the TR-FRET-based assay (
Figure 2A
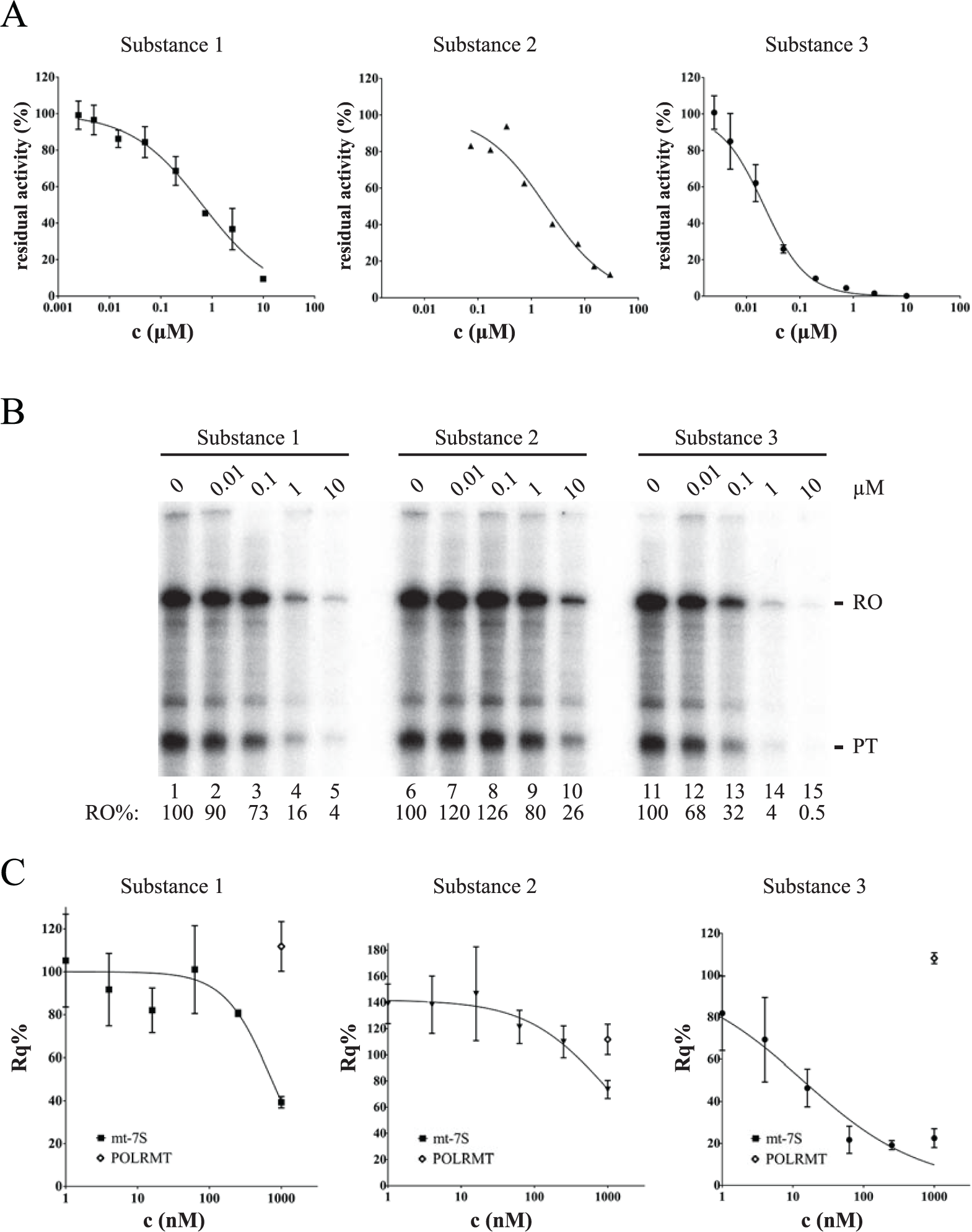
Effects of identified inhibitors on mitochondrial gene transcription in vitro and in cells.
As a final means of validation of the identified inhibitors of mitochondrial transcription, we assessed their cellular activity, measuring the dose-dependent inhibition of POLRMT-based transcription. Following incubation of HeLa cells with a dilution series of compounds for 4 h, RNA was extracted and the mitochondrial transcription activity of the cells determined via qRT-PCR. We used the mitochondrial 7S RNA to measure POLRMT activity ( Figure 2C ). mt-7S mRNA is a transcript of unknown function that is encoded proximal to the mitochondrial LSP. Human 7S RNA has a very short half-life (unpublished observation) and is therefore an ideal probe of de novo mitochondrial RNA synthesis. Established housekeeping-gene references were used to apply a comparative CT method (ΔΔCT) for analysis. In addition, we also monitored the abundance of the POLRMT mRNA itself to rule out that our results were influenced by transcriptional regulation of the nuclear-encoded polymerase gene. Our analysis yielded a group of 55 validated inhibitors, which displayed activities with IC50 values from 1 µM down to double-digit nanomolar in biochemical and cellular assays and which are selective for POLRMT. The identified inhibitors could be clustered into distinct structural groups with only a few singletons (data not shown). Moreover, all of the identified structure motives were novel, have never been reported in the context of polymerase inhibition, and were nonnucleotide analogs that should provide alternative mechanisms of action, compared to compounds targeting (e.g.) viral RNA-polymerases).
Recapitulating the outcome of our validation assay cascade, we observed excellent correspondence among the different assay formats used, and, in particular, we observed that the developed high-throughput assay format could be benchmarked as very predictive and robust. As an example, we here provide a compilation of the dose-dependent inhibition of mitochondrial transcription as measured using the newly developed TR-FRET assay format, a classical in vitro transcription assay with radioactive nucleotides, and qRT-PCR for three substances that were originally identified during HTS ( Figure 2A–2C ). We chose Substance 1 and Substance 3 as representatives of the same structural scaffold, and Substance 2 from an alternative structure group, to provide evidence of a broad range of inhibitory activity within one hit cluster and among different structural families. Thus, we determined IC50 values in the new, homogeneous biochemical assay format as 0.65 µM, 1.7 µM, and 21 nM, for Substance 1, Substance 2, and Substance 3, respectively ( Figure 2A ). The different activities of these compounds are clearly reflected in classical in vitro transcription assays with radioactive nucleotides ( Figure 2B ), in which significant inhibition to about half the signal could already be observed between 0.01 and 0.1 µM of Substance 3. Likewise, the lower activity and, importantly, rank order of Substance 1 and Substance 2 in biochemical homogeneous and blot-based assay formats were found to correspond to each other. To ensure that the activities observed in cell-based qRT-PCR were unbiased from the biophysical properties of the three inhibitors under scrutiny, we assessed their ability to permeate a parallel artificial membrane, as described in Ref. 14 , and determined very closely comparable values (60–70% flux). Hence, the observed cellular IC50 values of 0.73 µM, 0.98 µM, and 14 nM for Substance 1, Substance 2, and Substance 3, respectively, can be attributed to the inhibitors’ different activities on POLRMT within the mitochondria alone ( Figure 2C ). Moreover, the rank order of activities of the different inhibitors in the developed homogeneous biochemical assay format is in clear correlation, and the range of activities of the individual substances corresponds well with those of the cell-based qRT-PCR assay. Taken together, the newly developed assay technology enabled the discovery of valid inhibitors of POLRMT, and the used combination of assays yielded a consistent profile of the identified compounds.
All the inhibitors identified here were nonnucleotide analogs, but our HTP assay could potentially also be used to predict adverse side effects of NIs used to treat viral infections. These NIs are often administrated throughout prolonged periods of time (12–48 weeks) and may cause mitochondrial toxicity during the duration of the treatment. 4 To investigate the usefulness of our assay for this purpose, we analyzed a number of well-characterized NIs. These included the triphosphorylated forms of 2′ME/OH-GTP (INX-189), 2′ME/F-GTP (PSI-938), and 2′ME/OH-O-UTP (VX-135), which all have been shown to cause toxicity in humans ( Figure 3A ).1–3,15 Our analysis demonstrated that the analyzed NIs all inhibited POLRMT with an IC50 ranging from 112 to 480 µM in the new TR-FRET-based assay format. In contrast, we did not observe a similar inhibitory effect of the clinically approved compound 2′ME/F-UTP (sofosbuvir) ( Figure 3B ). These observations were verified in the orthogonal biochemical assay system, in which 2′ME/OH-O-UTP inhibited mitochondrial transcription, whereas 2′ME/F-UTP failed to affect transcription even at very high final concentrations (200 µM) ( Figure 3C ).
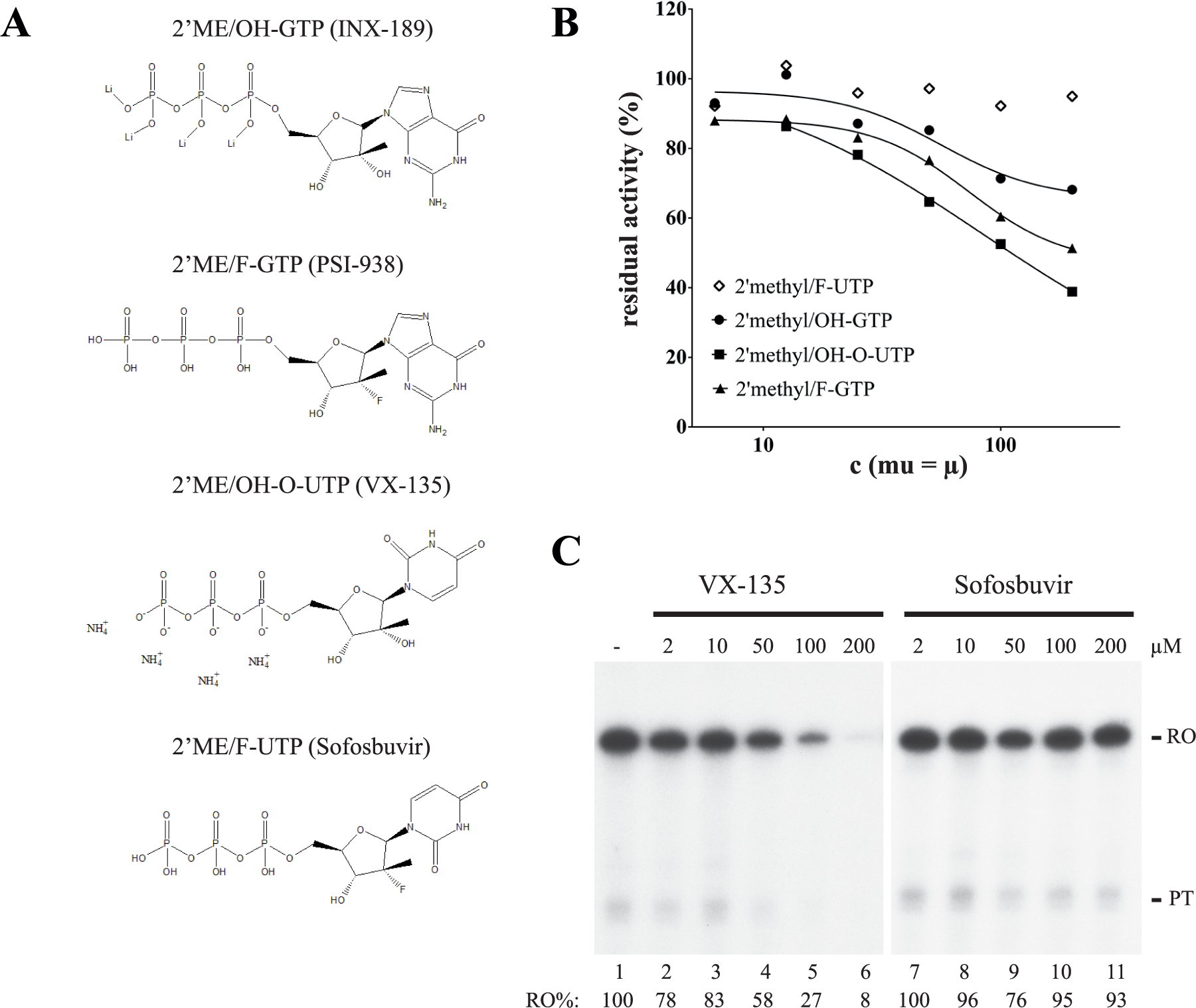
Effects of nucleoside inhibitors on mitochondrial transcription. (
Discussion
We here present the development of a robust method that can be used to measure RNA polymerase product formation by means of highly specific binding of DNA probes and, thus, generation of a proximity-based TR-FRET signal. This novel technology facilitated the establishment of homogeneous assay formats with working volumes of <10 µL. The method was amenable to the use of 1536-well microtiter plates, with high signal-to-background ratio and very robust assay performance and reliability (z’ >0.8). The assay has been tested and used for the identification of small-molecule modulators of mitochondrial transcription. The specific design of the detection method is conveniently adaptable to varying spectroscopic detection technologies and different DNA and RNA polymerases due to the use of readily available custom DNA synthesis. The assay can thus be used to assess selectivity of inhibitors against RNA polymerase from different organisms in a high-throughput format.
Recent reports suggest that inhibition of mitochondrial gene expression may be of therapeutic value in the treatment of certain forms of cancer (e.g., acute myeloid leukemia).16–17 Based on the obtained results and chemical evaluation, our validated inhibitors therefore provide promising starting points for a follow-up medicinal chemistry optimization program to generate druglike leads that are suitable for further development.
For a long time, it was believed that cancer cells did not need functional mitochondria but instead relied on aerobic glycolysis for production of ATP and building blocks (Warburg effect). Later studies have complicated this simple view, however, and it is now clear that tumors are less uniform than previously anticipated and contain a subset of cells that is strictly dependent on oxidative phosphorylation (OXPHOS). In fact, a number of reports have suggested that upregulation of OXPHOS may be a selective vulnerability in cancer stem cells and drug-resistant cancer cells, and in line with these observations chemical inhibitors of respiration can be used to eradicate cancer.18–19 Unfortunately, available inhibitors of respiration have narrow therapeutic indices and are for laboratory use only (e.g., rotenone). As an alternative, respiration can also be inhibited indirectly by inhibition of mitochondrial biosynthesis. The mitochondrial genome encodes key components of the OXPHOS system, and reduction of mtDNA gene expression may therefore cause a reduction in cellular respiration. As a proof of principle, the antibiotic tigecycline, which inhibits mitochondrial translation, was identified in a screen for compounds that specifically inhibited growth of OXPHOS-dependent leukemia cells. 16 Furthermore, a 2-C-methyladenosine (2-CM), which functions as a chain terminator of mitochondrial transcription, was shown to decrease acute myeloid leukemia cell growth in a mouse xenograft model. 17 The inhibitors identified here may be useful in further studies for effects on those specific forms of drug-resistant cancers, including lymphomas and melanomas, which are characterized by increased mitochondrial respiration. 18
NIs that affect mitochondrial gene transcription, by contrast, can also produce adverse side effects in vivo, even if these effects are often first seen after prolonged treatment. In previous studies, the ability of POLRMT to use a specific NI has been determined using RNA-primed DNA templates, which can monitor the incorporation of the first complementary nucleotide.4,20 In these assays, the fraction of primer extended after a defined time corresponds to the ability of POLRMT to use the specific NI. Even if this assay has proven useful, it differs quite substantially from the in vivo situation. The substrate used is single-stranded, whereas dsDNA is the normal, in vivo template for POLRMT. In fact, POLRMT is a very poor polymerase on single-stranded DNA (ssDNA) templates. Whereas POLRMT can synthesize long stretches of RNA on a dsDNA template, it is nonprocessive on ssDNA and transcription ceases after 25–75 nt. 21 Furthermore, a single nucleotide incorporation assay does not address the effect of NIs on transcription initiation, which has been shown to be very sensitive to changes in nucleotide concentrations. 22 Our novel method can therefore be useful for predicting effects on mitochondrial gene transcription during the development of NIs with antiviral activity.
Footnotes
Acknowledgements
The authors thank T. Hegendörfer, B. Klebl, and P. Nussbaumer for helpful discussions and critical reading of the manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: NGL, CMG, MF, and TB have a patent PCT/EP2016/062198 on the described method pending.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work by NGL and TB was supported by the Max-Planck Society; and MF and CMG were supported by the Swedish Cancer Society.