
Abstract
Epigenetic gene regulation is a critical process controlling differentiation and development, the malfunction of which may underpin a variety of diseases. In this article, we review the current landscape of small-molecule epigenetic modulators including drugs on the market, key compounds in clinical trials, and chemical probes being used in epigenetic mechanistic studies. Hit identification strategies for the discovery of small-molecule epigenetic modulators are summarized with respect to writers, erasers, and readers of histone marks. Perspectives are provided on opportunities for new hit discovery approaches, some of which may define the next generation of therapeutic intervention strategies for epigenetic processes.
Keywords
Introduction
Drug discovery at most pharmaceutical companies is transforming on many fronts, with a scrutinization and revamping from early discovery through to the preclinical and clinical arena. As drugs lose patent protection and R&D productivity is significantly challenged, this widespread overhaul sets out to change the blockbuster strategy that prosecuted classical, well-understood drug targets to a more entrepreneurial and collaborative approach encompassing novel targets and disease pathways, many of which are associated with much smaller patient populations.1–3 This overhaul counters some perceptions that the so-called “druggable genome” comprises only a limited number of targets and that the majority of tractable small-molecule drug targets have already been uncovered.2–5 A major focus of the past two decades of drug discovery has been on the connection of genetic mutations to diseases, catapulted by the ability to rapidly sequence and study the entire human genome. However, this approach has met with limited success. Many in the academic and industrial biomedical science communities are contending that the next wave of medicines and druggable targets will come from more complex biological areas and from potential root causes of diseases previously unknown, such as pathways that control protein stability, stem cell origins of disease, and so forth. One such fast-growing area for possible therapeutic intervention is epigenetics, which embodies heritable factors beyond the DNA sequence that control the expression of the genetic code.
Although the epigenetics field is still in its pioneering stages, it has been well reviewed. Recent review articles describe advances in our understanding of the components and processes that modulate the conformational state and dynamics of chromatin.6–11 These modulatory processes control accessibility of transcriptional machinery to genes and play an important role in gene transcription states such as silencing, repression, activation, and elongation. Increasing evidence in the literature has indicated that epigenetic regulation mechanisms act in concert to set up a specific and sustained chromatin state under physiological conditions, which can change during development or under disease conditions. These states include euchromatin for active transcription, poised states with basal levels of expression for further transcription regulation (activation/repression), or heterochromatin wherein transcription is silenced either permanently or conditionally. All factors of the chromatin (i.e., DNA, RNA, and proteins/histones) appear to play a role in the process. A number of key components governing chromatin dynamics, illustrated in Figure 1 , are being actively explored by academic groups and have the potential to become targets for disease modification strategies in the future; these components include DNA methylation at CpG islands, 12 noncoding RNA molecules,13,14 chromatin-remodeling complexes, 7 histone-modifying enzymes (histone “writers” and “erasers”), and proteins that specifically recognize and then translate the histone marks (histone “readers”). 6 Interplay among these epigenetic components is complex and not fully understood but is nonetheless essential to normal processes such as development and cell differentiation. Misregulation of the histone code and chromatin dynamics contributes to a variety of human diseases, including cancer, metabolic and cardiovascular diseases, and mental retardation.9,15–18
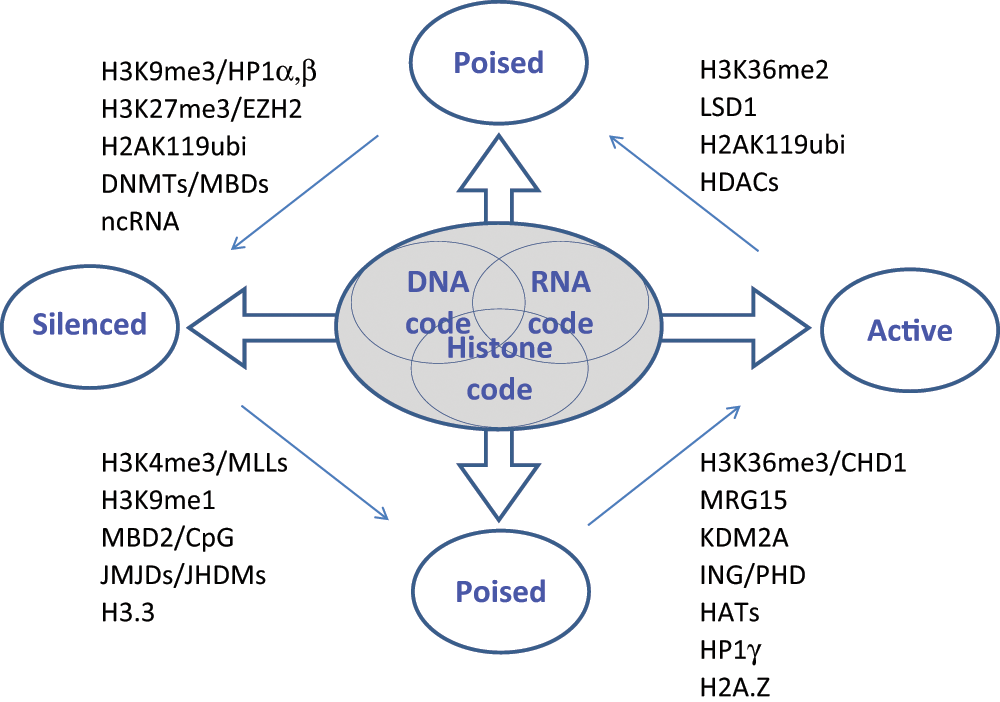
Interplay of the DNA code, the RNA code, and the histone code in epigenetic regulation of transcription. The figure depicts the complexity of epigenetic transcription regulation, wherein multiple factors contribute to the set up of a defined chromatin state. Representative proteins and modifications are shown for roles in transition among various transcriptional statuses. The histone code refers to the writing, erasing, and reading of histone marks as defined by Allis and colleagues. 6 The DNA code refers to promoter CpG methylation and accessibility of promoter elements mediated by nucleosome positioning. The RNA code refers to the roles of noncoding RNAs in setting up the chromatin structure.
In this article, we will review the current state of small-molecule epigenetic drugs on the market, chemical tool compounds (chemical probes) for selected epigenetic processes, and the various hit identification approaches being used for epigenetic targets and pathways. We also provide some perspectives on emerging and potentially promising drug discovery strategies based on targeting epigenetic systems.
The Current Landscape of Chemical Modulators of Epigenetic Proteins
Efforts toward the identification of chemical probes and chemotherapeutic agents modulating epigenetic processes have been in progress for more than 40 years, starting with the discovery and characterization of the DNA methyltransferase (DNMT) inhibitor 5-azacytidine.19,20 Despite many years of effort, there exist only four drugs approved by the Food and Drug Administration on the market that specifically target epigenetic regulatory proteins ( Table 1 ). Given the myriad biological and disease processes under epigenetic control, proteins associated with modulating chromatin structure/function and gene expression patterns represent an attractive, albeit challenging, class of drug targets for many therapeutic areas. 21 Thus far, DNA methylation and histone readers, writers, and erasers are considered the most tractable drug targets in epigenetics, yet comprise hundreds of proteins previously unknown or largely unaccounted for during the druggable genome debate in the past decade (www.thesgc.org/resources/histone_tails/). 22 In just the past few years, both industrial and academic groups have initiated a number of efforts to discover novel compounds that interact with these systems via screening and rational design approaches. This has resulted in significant advances in the development of robust assays and reagents.23–29 Even though lead discovery efforts against many classes of epigenetic targets have been initiated only relatively recently, many have already yielded promising compounds (e.g., Table 2 ), suggesting broad tractability for drug discovery and therefore tremendous future potential. Published inhibitors for each of the main epigenetic protein target classes are summarized in Table 1 , with selected cases further elaborated below.
Summary of Major Classes of Epigenetic Modulators Reported in the Literature
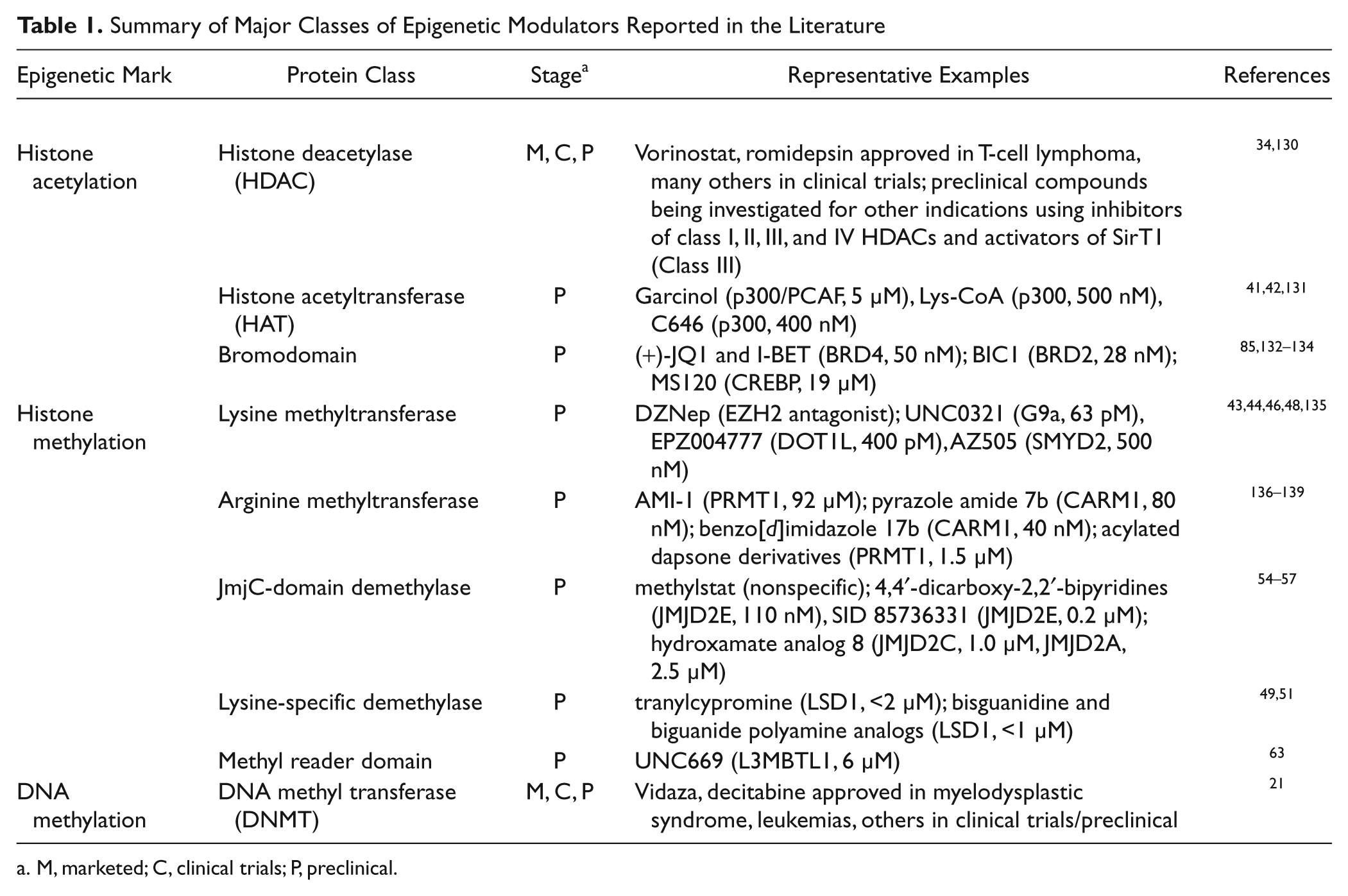
M, marketed; C, clinical trials; P, preclinical.
Summary of Probe Discovery Strategies and Target Exemplars
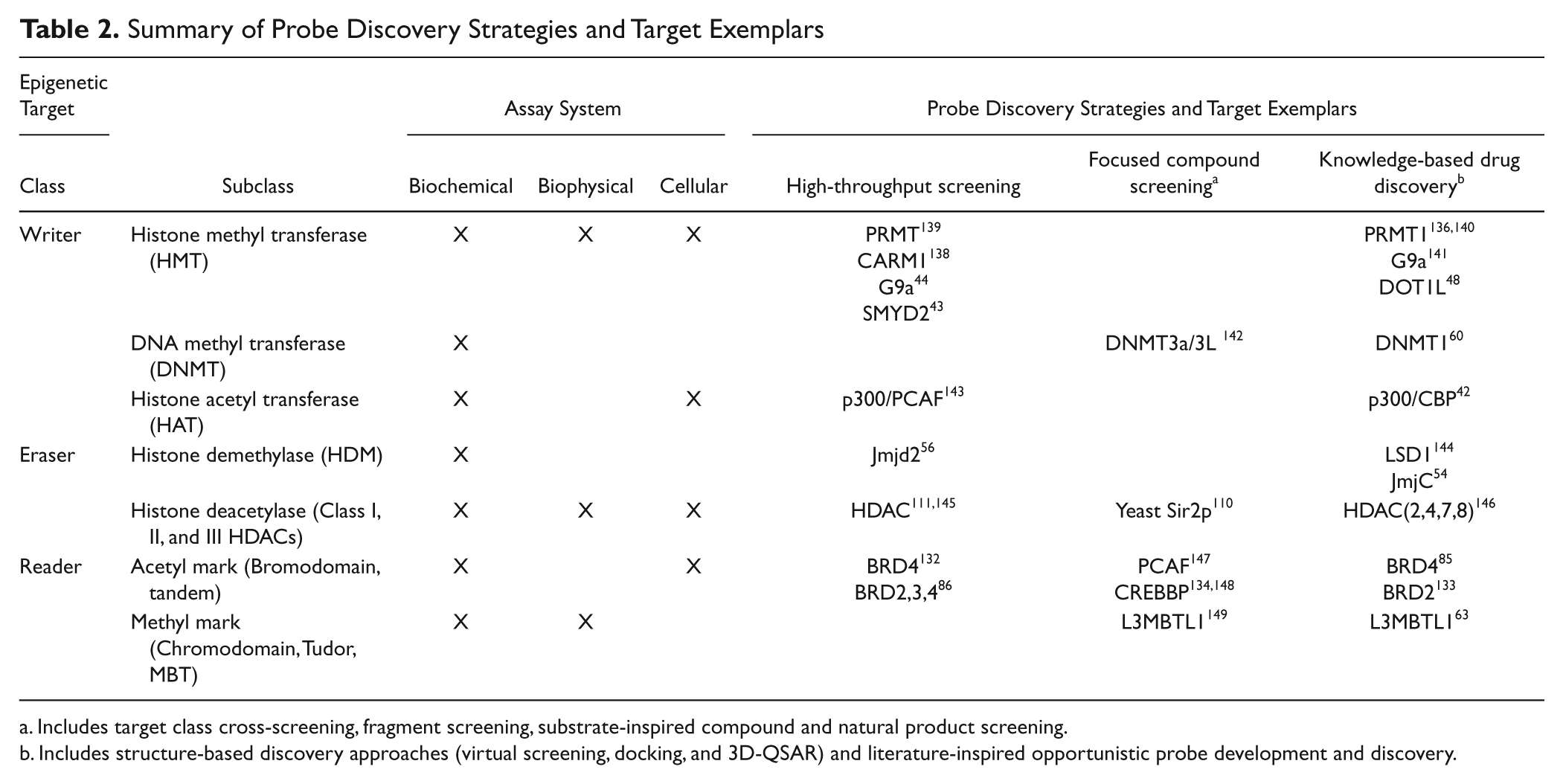
Includes target class cross-screening, fragment screening, substrate-inspired compound and natural product screening.
Includes structure-based discovery approaches (virtual screening, docking, and 3D-QSAR) and literature-inspired opportunistic probe development and discovery.
Modulators of Histone Acetylation
The most extensively studied histone modification is N-terminal histone tail lysine acetylation, where neutralization of the side chain’s positive charge with acetyl transfer results in decreased association with the positively charged DNA backbone. This decreased association leads to a more open and trancriptionally active form of chromatin. Acetylated lysine residues also serve as molecular recognition marks for the recruitment of proteins that further modify chromatin structure.30–32 The potential to affect a disease state upon modulation of histone acetylation has been validated by the pharmacological inhibition of histone deacetylase (HDAC) enzymes, resulting in reactivation of aberrantly silenced genes in cancer, including the cyclin-dependent kinase p21 (WAF1). 33
There are currently two HDAC inhibitors approved for treatment of cutaneous T-cell lymphoma. 34 The precise mechanism of HDAC inhibitors is uncertain, however, and the cytotoxic and antitumor effects of the inhibitors Vorinostat (SAHA) and romidepsin (Istodax) may involve changes in acetylation of nonhistone proteins important in signaling and cell growth, regulation of STAT transcription factors, and generation of reactive oxygen species, in addition to direct effects at promoter regions due to changes in histone acetylation.35,36 However, these inhibitors are not very specific, nor selective among the HDAC classes. Multiple HDAC inhibitors encompassing a range of chemotypes are in clinical trials as single agents or in combination with radiosensitization, protease inhibitors, DNMT inhibitors, and other drugs for the treatment of multiple cancers. Interest in developing class- and isoform-selective HDAC inhibitors is growing, 37 and it is anticipated that it may be possible to reduce certain toxicities and increase efficacy by targeting the relevant isoform(s) in a particular indication. 38
Despite the attention given to targeting HDACs in cancer treatment, the histone acetyltransferase (HAT) enzymes that catalyze transfer of acetyl groups to lysine residues are less understood. Many of these enzymes, including p300 and members of the MYST family of HAT, are associated with cancer as well as inflammation.39,40 Like the HDACs, many HAT enzymes use nonhistone substrates that may contribute to their downstream cellular effects. Several inhibitors of HAT enzymes have been discovered, including the natural products garcinol, curcumin and its cinnamoyl derivatives, and anacardic acid. Improved potency and selectivity were achieved with synthetic conjugates of coenzyme A with either lysine (Lys-CoA, p300 IC50 = 0.5 µM) or histone peptide (H3-Lys-CoA, PCAF IC50 = 0.3 µM), although pharmacokinetic properties limit their use as tool compounds. 41 Cellular decreases in histone acetylation levels became possible with the identification of compound C646 through virtual screening of the p300 HAT. 42 This and other small-molecule chemical probes will be important in elucidating the biological function and disease association of HAT enzymes.
Modulators of Histone Methylation
Following the discovery of the reversible nature of histone methylation and its association with a range of human diseases, histone tail methylation of both lysine and arginine residues has received significant attention. 18 Histone methyltransferase (HMT) and histone demethylase (HDM) enzymes exert tight control over the complex code written by these modifications, wherein both the site and degree of methylation (mono-, di-, or tri-) can modulate transcription. More than 50 HMTs have been discovered in the past decade, and ample data suggest that some of these are attractive drug targets. 18 Although no marketed drugs exist to modulate HMT activity, several small-molecule probes have been recently identified and used to examine the biological roles of these histone-modifying enzymes ( Table 1 ).
Recent high-throughput screens (HTS) have identified tractable hits for G9a and G9a-like protein (GLP), two structurally related lysine methyltransferases, for the arginine methyltransferase CARM1 and for the lysine methyltransferase SMYD2. 43 Crystal structures of these methyltransferases bound to their respective small-molecule inhibitors have been deposited in the Protein Data Bank (PDB).44–47 These structures provided insight on how the compounds bind and inhibit catalytic activity ( Figure 2 ). Furthermore, the structures have guided the synthesis of analogs with improved potency.46,47 The G9a/GLP inhibitors have reported selectivity versus related lysine methyltransferases such as SUV39H2, SET7/9, and SET8,44,46 and the CARM1 inhibitors have selectivity versus the structurally related arginine methyltransferases PRMT1 and PRMT3. 47 The potent and specific DOT1L inhibitor EPZ004777, derived from molecular docking of SAM mimetics at the active site of DOT1L, was shown to specifically block cellular H3K79 methylation and selectively kill cells bearing mixed lineage leukemia (MLL) translocations. 48
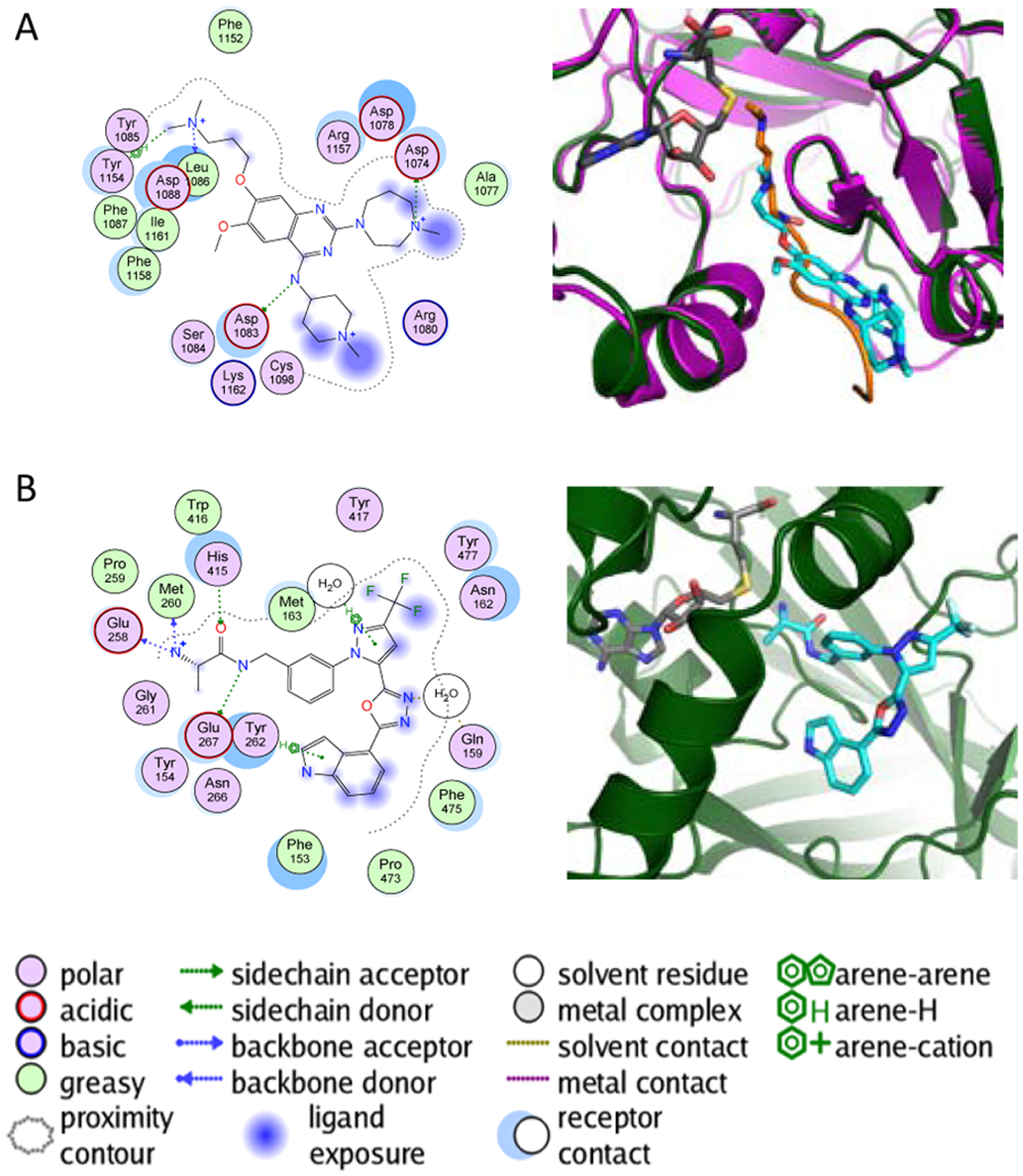
Substrate-competitive inhibitors of methyltransferases. (
Chemical probes of HDMs have also been developed to determine biological activity and validate these enzymes as potential therapeutic targets ( Table 1 ). The lysine-specific demethylase LSD1, an FAD-dependent oxidase, is inhibited by compounds whose activity was discovered on the basis of mechanistic similarity to monoamine and polyamine oxidase enzymes. Following the discovery that the monoamine oxidase (MAO) inhibitor tranylcypromine inhibits LSD1 with an IC50 less than 2 µM, 49 derivatives of tranylcypromine have been developed with improved potency, H3K4me2 cellular activity, and LSD1 selectivity. 50 Other MAO inhibitors, pargyline and phenelzine, are themselves weak inhibitors of LSD1, but peptide analogs of these compounds show LSD1-selective inhibition. Polyamine-based LSD1 inhibitors have also been developed, including (bis)guanidines, (bis)thioureas, and oligamines that show cellular effects on histone methylation and transcriptional activation of aberrantly silenced genes.51–53 Members of the other family of HDMs, the Fe(II) and 2-oxoglutarate (2-OG)–dependent JmjC-domain demethylases, are inhibited by N-oxalylglycine (NOG) and its derivatives, which are small-molecule analogs of 2-OG.54–57 Additional small-molecule probes have been identified for JmjC-domain demethylases including bipyridines,55,58 8-hydroxyquinolines, 56 and the bisubstrate analog Methylstat. 54 Methylstat and a hydroxamate analog of NOG have shown JMJD-dependent inhibition of cancer cell lines.54,57
Modulators of DNA Methylation
In addition to methylation of histone tails, methylation of DNA at CpG islands of promoter regions is an important mechanism of epigenetic regulation. DNMT activity is generally associated with epigenetic silencing, so that modulation of DNMTs is an attractive approach to reactivation of silenced tumor suppressors. 21 Approved DNMT modulators 5-azacytidine (Vidaza) and 5-aza-2′-deoxcytidine (Dacogen) are nucleoside analogs used to treat myelodysplastic syndrome (MDS). The mechanism of action involves incorporation of deoxynucleotide triphosphates into DNA, acting as suicide substrates of DNMTs, thereby covalently trapping the enzyme on the DNA strand. Degradation and depletion of DNMTs lead to reactivation of silenced genes, such as E cadherin, through decreased promoter methylation. Changes in DNA methylation are not strongly linked to therapeutic efficacy, however, and clinical responses observed may be due in part to cytotoxicity of bulky, trapped DNMT/DNA complexes. 59 Multiple clinical trials are under way to expand indications of nucleoside analogs for the treatment of various hematological and solid tumors. S-110, a prodrug of 5-aza-2′-deoxcytidine, is also in clinical trials for the treatment of MDS and acute myeloid leukemia. Several nonnucleoside DNMT inhibitors are undergoing preclinical testing, including the ECGC polyphenol, RG108 DNMT1 inhibitor, and MG98 antisense oligonucleotide. 60
Modulators of Histone Code Readers
Histone code readers for acetyl marks and methyl marks are the two major classes of proteins that function to recognize and interpret the histone code. Proteins that recognize acetylated histones contain one or more of the structurally conserved bromodomains, whereas proteins that recognize methylated histones contain one or more reader domains including the chromodomain, the tudor domain, the MBT domain, the PHD domain, and/or the WD40 domain. 30 Protein structures for many of the above classes of histone code readers have been solved by either x-ray crystallography or nuclear magnetic resonance in recent years and deposited in the Protein Data Bank (PDB). Structural elucidation of histone code readers has enhanced our understanding of the important binding interactions between the reader protein and modified histone marks. This knowledge has facilitated the identification and development of potential small-molecule inhibitors of the histone reader-mark interaction. For example, the 3,5-dimethylisoxazole moiety was identified as an effective acetyl-lysine mimic, and a derivative of this was identified as a selective and potent inhibitor for the bromodomain proteins BRD2 and BRD4. 61 Methyl-lysine readers need to accommodate a cationic moiety with varying degrees of amino acid methylation. Recognition by the protein of these lysine methylation states is achieved by caging the methylammonium moiety with aromatic residues. These aromatic cages interact with the quaternary ammonium functionality primarily through cation-π-type interactions, and recognition of lower methylation states involves hydrogen bonding of the protonated amine, as exemplified by the crystal structure of L3MBTL1 complexed with H4K20Me2 (PDB accession code 2RJF; Fig. 3A ). More details are included in a review by Taverna and colleagues. 30
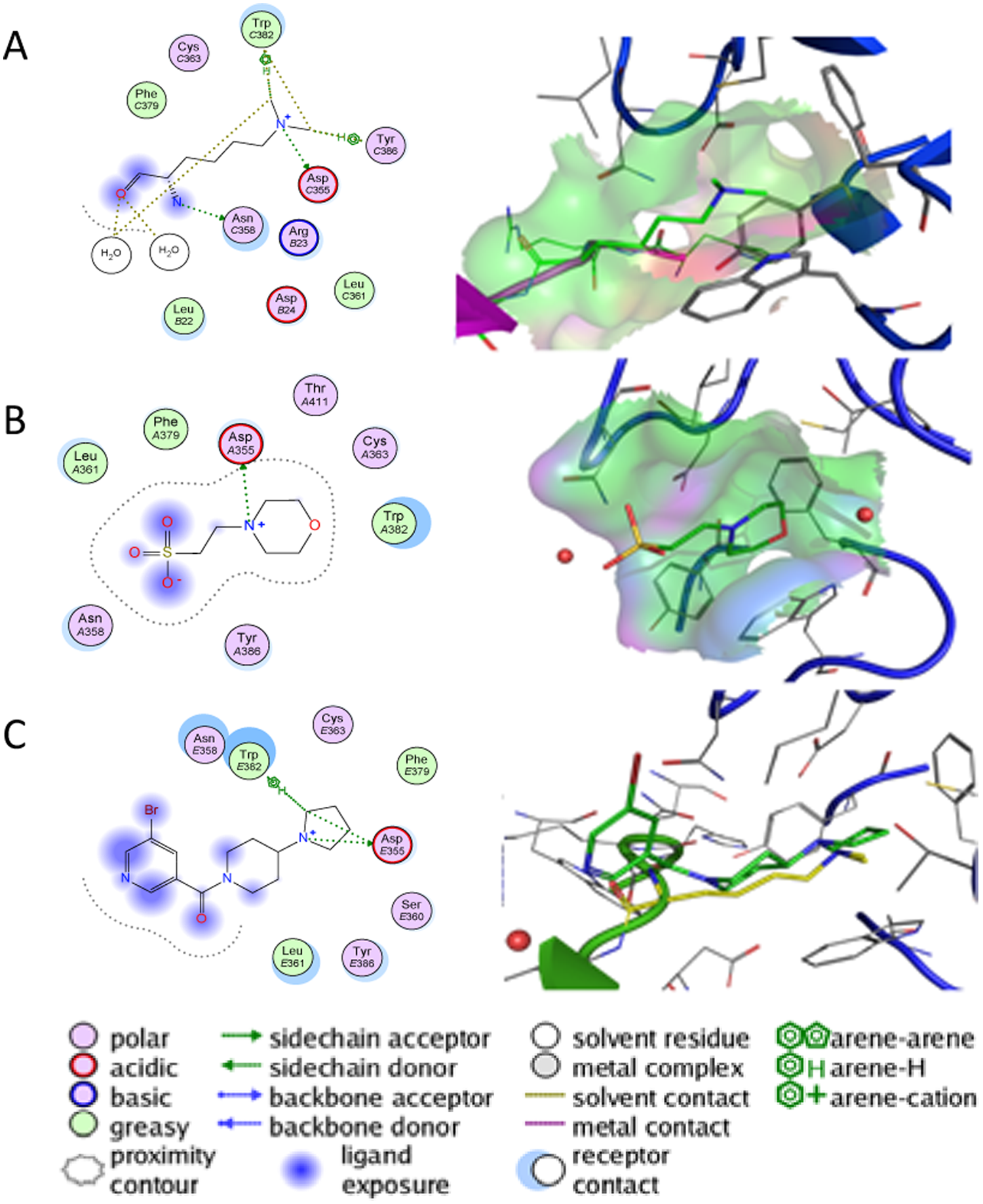
Compound interaction with the Royal family protein L3MBTL1. (
Campagna-Slater and Schapira 62 reported on the use of pharmacophore queries of the PDB, built on aromatic cages, to help identify potential antagonists of histone code readers. A large number of aromatic cages were retrieved by the searches from proteins unrelated to the histone code readers employed to build the pharmacophore query. Examples of ligands bound to aromatic cages include quaternary ammonium and sulfonium analogs, tertiary, secondary and primary amines, imines, aromatic rings, and other miscellaneous compounds such as aliphatic rings, pyridine, and N-oxides. Consistent with the results of these pharmacophore searches, coordinates for the methyl reader L3MBTL1 in complex with 2(N-morpholino)-ethanesulfonic acid (MES), a common buffer additive, had been solved showing the morpholine ring bound to the aromatic cage ( Fig. 3B ). Recently, small-molecule ligands for L3MBTL1 that antagonize native histone binding have been described. 63 The compounds were obtained by design of peptidomimetic structures based on the co-crystal structure of L3MBTL1 complexed with H4K20Me2. Replacement of the methyl-lysine amine moiety with simple amines led to the observation that a pyrrolidine analog had better binding affinity for the protein than did mono- and di-methyl-lysine modifications. Through a series of structural modifications, based on virtual screens and rational design, a nicotinamide analog was found that antagonizes the binding of histone peptide in both AlphaScreen and ITC assays. The compound was crystallized in complex with L3MBTL1 and binds in the same pocket as H4L20Me2, with the pyrrolidine ring accommodated in the aromatic cage ( Fig. 3C ).
Emerging Hit Identification Strategies for Epigenetic Targets
As summarized above, hit discovery strategies for epigenetic targets so far have mainly focused on antagonizing catalytic activities of histone/DNA–modifying enzymes and have been considered successful in obtaining promising compounds ( Tables 1 and 2 ). As our understanding of epigenetic processes and underlying cellular biochemistry improves, and as we make use of the initial discoveries of chemical probes, customized and integrated hit-identification strategies for specific epigenetic targets or pathways will emerge over the next few years. This section focuses on some of the promising approaches, with an emphasis on those targeting the function of epigenetic protein complexes.
Tailoring Compound Screening Collections for Epigenetic Targets
Efforts to discover novel chemical probes that modulate epigenetic targets have benefited from HTS as an integral component of the hit identification strategy ( Table 2 ). An outstanding question, which is currently being answered via ongoing screening activities, is whether the routine discovery of chemical probes that target epigenetic proteins can be obtained from general HTS diversity collections or whether specific efforts need to be applied to build collections that will be effective for these targets. Preliminary indications suggest that epigenetic targets may have similar tractability in HTS to more “classical” drug targets, such as G-protein–coupled receptors (GPCRs), proteases, and kinases. Nonetheless, there is considerable ongoing effort to build focused compound libraries that may be enriched for compounds that hit epigenetic targets. For example, the SET domain-containing histone methyltransferases have a conserved active site with an S-adenosyl methionine (SAM) binding pocket and a variable nucleosome binding site, which is analogous to the Ser/Thr/Tyr protein kinases with a highly conserved ATP binding pocket. Thus, the same hit identification and medicinal chemistry approaches that have led to the development of specific and clinically efficacious therapeutics targeting kinases could, in principle, be applied to histone methyl transferases. Building SAM mimetic compound libraries to screen both histone and DNA methyltransferases may provide an entry point for selective SAM-competitive inhibitors. Targeting the histone-binding site, on the other hand, will be aided by the availability of crystal structures and modern computational chemistry tools to define the structural tolerance and limitations of these binding pockets.
Alternative Hit Identification Methods
In addition to HTS and the use of focused compound libraries, a range of other methods may have utility for targeting epigenetic proteins for hit identification. One example is Encoded Library Technology (ELT), which allows very large numbers of diverse compounds to be screened for their ability to bind to a target via a relatively facile process. 64 ELT libraries consist of compounds tagged with unique DNA sequences that enable identification of specific binders through PCR amplification and deep sequencing of the DNA barcode tags. This approach has been successful for the discovery of inhibitors of Aurora A and p38 MAP kinases that were shown to bind to the ATP site. 64 As relatively few chemotypes have been identified for the multitude of epigenetic targets, conventional high-throughput and virtual screening approaches may be lacking in chemical space for epigenetic targets. DNA-encoded libraries composed of thousands to billions of compounds can be rapidly synthesized through iterative splitting and pooling of chemical reactions with stepwise DNA tagging, and these can be screened in parallel because of the sensitivity of high-throughput sequencing. Selection conditions may also be easily modified to address relevant questions; for example, selections may incorporate multiprotein complexes or tool compounds (e.g., SAM) to identify ligands of specific binding sites. As this method requires a very small amount of protein to perform library selections (10’s of micrograms), it has potential to be used on a panel of epigenetic proteins as a rapid method to scan for potential hits and to assess chemical tractability (J. Gross, unpublished).
Another method that appears to have clear potential utility for the discovery of epigenetic modulator compounds is fragment-based drug discovery. 65 This method is based on the detection and then elaboration of small fragments (e.g., MW <200 Da) that bind efficiently to drug targets that are amenable to the use of structural methods such as x-ray crystallography or nuclear magnetic resonance (NMR). Rapid progress is being made in protein structure studies for specific domains of epigenetic systems, which would seem to prime this area for future success.
Targeting Protein-Protein Interaction of Readers and Enablers
Protein-protein interactions (PPIs) underpin virtually all of the epigenetic mechanisms, including covalent histone modification and recognition, ATP-dependent chromatin remodeling, methylated CpG-mediated gene silencing, and RNA-mediated epigenetic regulation ( Fig. 1 ). The same challenges associated with interfering with PPIs in other target classes exist with protein-protein or protein-nucleosome interactions of epigenetic proteins, wherein a large binding energy exists via the coordinated action of multiple molecular interactions, including van der Waals contacts, hydrogen bonds, ionic bonds, hydrophobic interactions, and pi stacking interactions. PPI sites are typically large, flat, and shallow. In addition, they often are composed of multiple subsites and are relatively hydrophobic compared with typical small-molecule binding sites. Historically, PPIs have been deemed undruggable and have been generally avoided as targets for small-molecule probe/drug discovery. However, recent successes in probe discovery targeting PPIs have challenged this notion.66,67 Ideally, this can be achieved by the binding of a small molecule at hotspots, that is, interface residues that encapsulate the bulk of the free energy change on complex formation. Hot spots can be determined experimentally or computationally,68–70 and this information has been compiled in databases.71,72 For example, small-molecule inhibitors for MDM2/p53, cis-imidazolines, bind to MDM2 at its interface with p53 by mimicking the interactions made by three of its hotspot residues (L26, W23, F19). 73 PPI inhibitors with additional mechanisms have also been reported.66,67 A small allosteric inhibitor of TNFα destabilizes the active trimer by binding to the dimer and wedging the two subunits apart so the third subunit cannot bind. 74 In addition, virtual screening of the GTP-binding complex Arf1/ARNO found an inhibitor that binds Arf1 near its interface with ARNO, allowing the complex to form but impairing a conformational change required for its activation. 75
A variety of computational approaches have been used to predict the druggability of protein targets, 76 where druggability is defined as the ability of a protein pocket to bind a small molecule. For example, Fuller et al. 77 detected druggable binding sites at protein-protein interfaces using a small-molecule pocket-finding algorithm. 78 The authors noted that PPIs are characterized by several adjacent small pockets (6 ± 3 pockets with ~54 Å3 volume/pocket on average), in contrast to the one large pocket (~500 Å3 on average) found in typical protein-ligand interactions. Their study led to a strategy of simultaneously targeting multiple subsites of PPI targets. 77 In addition, Hajduk et al. 79 used the Druggability Index to prioritize targets by predicting their NMR screening hit rates using a model derived from polar and nonpolar surface area, surface, complexity and pocket dimensions. Another method, computational solvent mapping, 80 finds favorable interaction sites by moving small organic solvent groups around the protein surface, replicating experimental results. 81 Similarly, Seco et al. 82 predicted druggable binding sites by performing molecular dynamics simulation of the protein within an isopropanol-water box and identifying clusters of isopropanol as surrogates for druglike molecules. In contrast to the above-mentioned methods for targeting a binary complex, a study by Sugaya and Ikeda 83 led to a prediction that PPIs with multiple interacting partners and versatile functions are more likely to be druggable targets.
Protein targets that are predicted to be druggable for which a crystal structure is available can be used for virtual screening or structure-based rational design. A variety of epigenetic targets have been subjected to virtual screens, some followed by relevant biochemical, biophysical, and functional studies.60,84 The BET family bromodomain inhibitor (+)JQ1 was initially derived from molecular modeling of a binding pocket in an apo structure of bromodomain, 85 and the DOT1L inhibitor EPZ004777 was derived from docking SAM mimetics in the DOT1L active site. 48
The design of relevant assay and screening strategies targeting epigenetic PPI targets has been limited by the lack of a comprehensive understanding of the dynamic interactions that occur between chromatin and its readers or modifying enzymes or enzyme complexes in the disease state. However, steady advances have occurred in recent years with the characterization of specific molecular interactions between recognition domains and modified histone peptides. 11 The interactions between the surface groove or binding cleft of acetyl-lysine reader domains and the cognate-modified histone are predominantly stabilized via a network of water-mediated hydrogen bonds, with more specific hydrogen bonds to active site residues forming deeper in the pocket. Recently, a series of benzodiazepine analogs with high nanomolar affinity for the BET family of bromodomains have been identified. X-ray crystal structures of these compounds bound to the Brd2 and Brd4 bromodomains show that the two nitrogens of the 1,2,4-triazolyl ring of the benzodiazepine mimic the carbonyl moiety of the acetyl head group. 86 In addition to reading of a specific single histone mark, coordinated reading of multiple histone marks appears to be a common mechanism.10,87 For example, Zeng et al. 88 demonstrated that the tandem PHD finger of DPF3b functions cooperatively and is interdependent with the acetylation and methylation state of histone H3 tails to effectively control gene expression.
Targeting Allosteric Sites of Writers, Erasers, and Readers
Examples of allosteric inhibition of epigenetic targets are currently limited, even though allosteric modulation of kinases and GPCRs has yielded ligands with improved selectivity and chemical tractability.89–91 The regulatory mechanisms for several histone methyltransferases involve sites distal from the site of catalysis. The histone methyltransferase activity of EZH2, for example, requires complex formation with EED and SUZ12 to form the polycomb repressor complex 2 (PRC2). 92 Furthermore, PRC2 activity has been shown to be differentially regulated by the binding of EED to trimethylated histone tails; binding to H3K27me3 stimulates methyltransferase activity, whereas binding to H1K26me3 inhibits its activity on nucleosomes.93,94 Structural studies indicated that EED specifically binds to histone tails carrying trimethyl-lysines in an aromatic cage on the top face of its beta-propeller architecture ( Fig. 4A ), leading to allosteric activation of the PRC2 activity, whereas the opposite side of the EED beta-propeller structure binds to a 30-residue helical peptide from the N-terminus of EZH2.93–95 Disrupting the complex via specific interactions at critical regions of EED may represent a mechanism for allosterically inhibiting EZH2 activity. As a means to support further exploitation of this idea, a fluorescent polarization–based peptide displacement assay was recently developed for screening of compounds that disrupt the reader/substrate interaction between EED and H3K27me3. 96
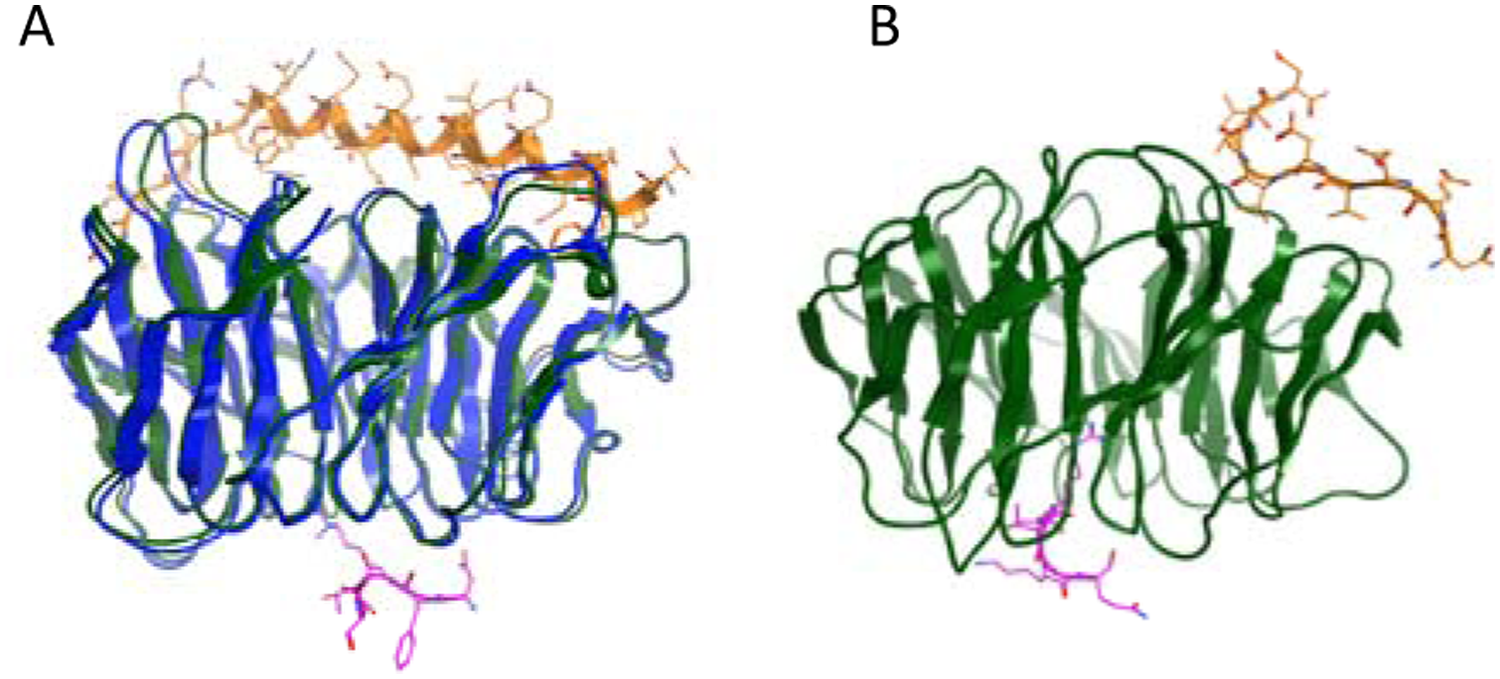
Protein-protein interactions for methyltransferase enablers. (
Along with EED, another structurally related protein, WDR5, also plays critical roles in coordinating formation and activity of multiprotein complexes. These WD-repeat proteins are characterized by seven WD40 repeats or β-propeller blades that are organized around a central cavity. WDR5 is a critical component of the SET1-family methyltransferase complexes. Like EED, the WDR5 protein has two binding faces. One side of the WDR5 protein has a rather deep arginine binding pocket that recognizes Arg-2 of histone H3 as well as Arg-3765 of the Win domain from MLL1.97–100 The opposite face of WDR5 binds the C-terminal tail of RbBP5, another WD-repeat protein. The structure of a ternary complex of the histone H3 tail peptide and the C-terminal tail of RbBP5 bound to WDR5 has been solved100,101 ( Fig. 4B ). The authors proposed a model of the WDR5, RbBP5, and MLL1 active complex, which suggests that WDR5 acts as a hub that secures MLL1 on one side and RbBP5 and its effector protein on the other.100,101 This model incorporates the available biochemical data indicating that WDR5 is necessary for full methyltransferase activity but is not sufficient to directly enhance activity in the absence of RbBP5. 101 As in the case of EED, targeting either of the binding faces of WDR5 with a small molecule or peptidomimetic may regulate MLL1 methyltransferase activity. A fluorescent polarization–based peptide displacement assay was also developed for screening of compounds that disrupt PPI between MLL and WDR5. 96
Another example of allosteric inhibition of an epigenetic enzyme was reported for DNMT1, in which the enzyme undergoes a slow relief from inhibition by unmethylated DNA as catalysis is initiated with the same molecule, resulting in a kinetic lag in product formation. 102 This unique mechanism can be exploited by exploring the chemical space around analogs of unmethylated DNA or nucleotides. As a further example, the histone demethylase LSD1 contains the functionally essential N-terminal SWIRM and Tower domains critical for protein stability and physiologically relevant interaction with its partner CoREST, respectively. 103 Reports have indicated that accessory proteins that interact with LSD1 serve a putative regulatory function by modulating its activity and substrate specificity.104–106 Although specific allosteric hotspots on the Tower domain have not been identified, Chen et al. 103 have postulated that CoREST binding to LSD1 may induce conformational changes that transform the site of catalysis into an active conformation. These findings present a unique opportunity for chemotherapeutically tuning LSD1 activity with finer control (i.e., differentially modulating demethylase activity of dimethylated versus monomethylated histones) than simply inhibiting all catalytic activity.
Targeting Epigenetic Pathways via Phenotypic Screens
Although most of the hit discovery approaches for epigenetic modulation thus far in production involve a very reductionist perspective (i.e., single-target design or screening campaigns), a more holistic approach may be needed for effective hit identification to address the complexities of epigenetic processes. Cell-based phenotypic screens employing physiologically relevant cells provide advantages over single target-based screens in that a wider target space such as signaling pathways, metabolic networks, and the entire proteome/epigenome 107 is exposed to the compound-screening collection. Vorinostat (SAHA) was discovered from a focused screen that induced cell differentiation with a series of bishydroxamic acids 108 and was later identified as an HDAC inhibitor. 109 A cell-based HTS for upregulation of ApoAI transcription resulted in the identification of compounds that disrupt PPIs between bromodomain proteins and acetylated histones. 86 A number of cell-based screens for epigenetic modulators have also been reported, including those for SIRT2 inhibitors, 110 HDAC inhibitors, 111 and de-repression of E-cadherin. 112 In all three cases, cells harboring a silenced promoter-reporter construct were used to identify compounds that caused de-repression of the reporter. The study by Best and colleagues 111 used GFP-fusion protein as a reporter to facilitate a high-content fluorescence-based screen for HDAC inhibitors. Given the role of histone marks in controlling gene expression, it seems that screening and/or compound-profiling approaches based on measuring changes in sets of gene transcripts are likely to be adopted in the future.
Identification of the specific target(s) of a hit compound and hence the potential mode of action can be challenging, especially if the compound has a low target affinity or is relatively nonspecific. Two main chemoproteomic approaches have been applied to achieve this goal,113,114 one based on compound affinity chromatography coupled with mass spectrometry 113 and the other based on stable-isotope labeling by amino acids in cell culture (SILAC).115,116 A target deconvolution study for compounds that upregulated ApoAI led to identification of bromodomain proteins as the molecular targets. 86 In addition to aiding target deconvolution, such chemoproteomics approaches also provide powerful tools for compound profiling for specificity or off-target effects. 117 A study of a set of HDAC inhibitors enabled characterization of target specificity of individual compounds and allowed identification of new targets. 118 SILAC-based approaches have also been developed to identify protein/DNA interactions 119 and to profile protein posttranscriptional modifications (PTMs), including methylation and phosphorylation.120,121 With the use of an appropriate antibody for specific histone modification, 122 SILAC approaches could potentially be developed for epigenetic PTM profiling.
Concluding Remarks
Epigenetic processes are central to cell function and specialization, the potential for potent and specific modulators of these systems to affect biological processes is extremely exciting. Considerable effort is ongoing to find molecules for many classes of epigenetic targets, and early indications suggest that these systems may be tractable using conventional lead discovery approaches and libraries. However, drug discovery in the area is likely to be quite challenging, and many questions remain to be answered before epigenetic systems can be more widely exploited for therapeutic uses. For example, what are the mechanisms behind the malfunctioning of histone readers, writers, or erasers in diseases? How is specificity achieved? Of utmost importance is gaining a better understanding of the epigenetic regulatory mechanisms involved in development and specific diseases, which are critical for discerning the causal events and identification of druggable targets. For example, understanding how oncogenic pathways target histone/CpG-modifying enzymes to gene promoters in cancer will enable the design of appropriate strategies for assay development and screens. Epigenetic regulation of gene expression is delicately balanced by a combination of positive and negative epigenetic marks and associated proteins ( Fig. 1 ) that are influenced by cellular signaling pathways under specific physiological/pathological conditions. It is thus reasonable to assume that effective epigenetic modulation is likely to be achieved by combination therapy with compounds that each target a distinct mechanism 123 ( Table 1 ). Conversely, targeting multiprotein complexes with a distinct function could also be exploited. For example, the epigenetic code replication machinery, which controls epigenetic cell memory, may represent promising druggable targets. 124 For modulating complex diseases involving multiple mechanisms and pathways, more physiologically relevant approaches such as phenotypic-based or function-based screens may be more appropriate than the single-target strategies.125,126 It is also interesting to speculate whether cross-reactivity may occur between existing (non–epigenetic-targeted) drug candidates and epigenetic systems and what impact this has on understanding the safety and efficacy profile of those molecules.
Epigenetic regulation involving histone modifications usually affects a set of genes, and epigenetic changes are typically differentially regulated with respect to developmental stages and tissue specificities. A major challenge in epigenetic drug discovery lies in assessing the liability of potential therapeutic compounds. Although changes in epigenomic profiles upon compound treatment can be obtained, these data need interpretation as to what levels of changes are considered normal and what are adverse. This will in turn provide guidelines to devise appropriate safety assessment procedures to establish appropriate compound dose, route of administration, exposure time, and duration of treatment. 127 Furthermore, the long-term impact of epigenetic treatment, including transgenerational disease susceptibility, may need to be evaluated.128,129
With focused research in academic institutions and increased investment in industry, it is anticipated that novel mechanisms of epigenetic modulation and small-molecule probes will be discovered. With establishment of procedures to achieve proper compound efficacy and safety profiles, there is hope in developing additional successful therapeutic modulators of epigenetically driven diseases.
Footnotes
Acknowledgements
We thank Dr. Peter Tummino, Dr. Jason Witherington, Dr. Chun-Wa Chung, Dr. Marti Head, and Dr. Gordon McIntyre for critical reading of the article. We also thank Dr. Roland Annan for discussions on applications of SILAC-based technology.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
Abbreviations: ELT, encoded library technology; DNMT, DNA methyltransferase; GLP, G9a-like protein; HAT, histone acetyltransferase; HDAC, histone deacetylase; HDM, histone demethylase; HMT, histone methyltransferase; HTS, high-throughput screen; MAO, monoamine oxidase; MDS, myelodysplastic syndrome; MLL, mixed lineage leukemia; NMR, nuclear magnetic resonance; NOG, N-oxalyl glycine; PDB, protein data bank; PPI, protein-protein interaction; PTM, posttranslational modification; SAH, S-adenosyl homocysteine; SAM, S-adenosyl methionine; SILAC, stable-isotope labeling by amino acids in cell culture.