
Abstract
This article presents a microfluidic approach for the integration of the process of aptamer selection via systematic evolution of ligands by exponential enrichment (SELEX). The approach employs bead-based biochemical reactions in which affinity-selected target-binding oligonucleotides are electrokinetically transferred for amplification, while the amplification product is transferred back for affinity selection via pressure-driven fluid flow. The hybrid approach simplifies the device design and operation procedures by reduced pressure-driven flow control requirements and avoids the potentially deleterious exposure of targets to electric fields prior to and during affinity selection. In addition, bead-based reactions are used to achieve the on-chip coupling of affinity selection and amplification of target-binding oligonucleotides, thereby realizing on-chip loop closure and integration of the entire SELEX process without requiring offline procedures. The microfluidic approach is thus capable of closed-loop, multiround aptamer enrichment as demonstrated by selection of DNA aptamers against the protein immunoglobulin E with high affinity (KD = 12 nM) in a rapid manner (4 rounds in approximately 10 h).
Introduction
Aptamers are single-stranded oligonucleotides (typically 12–80 nucleotides long) that function as specific affinity receptors toward a broad spectrum of biological targets, including small molecules, 1 peptides, 2 amino acids,3,4 proteins,5,6 cells,7,8 viruses,9,10 and bacteria. 11 With advantages such as synthetic availability through an in vitro process, ease of coupling to other functional molecular groups,12,13 reversibility of binding, long-term stability, low immunogenicity, 14 low batch-to-batch variability, 15 and long shelf life, aptamers offer an attractive alternative to other affinity receptors, (e.g., antibodies16,17) and have broad applications to basic biological sciences, 18 drug discovery, 19 and clinical diagnostics and therapeutics.16,20
Aptamers are discovered from large randomized libraries of oligonucleotides through an in vitro process termed systematic evolution of ligands by exponential enrichment (SELEX).17,21,22 SELEX involves iterative rounds of affinity selection against a target of interest, as well as amplification via PCR, of target-binding oligonucleotides from large randomized libraries (up to 1014 members). 23 Conventional SELEX platforms require extensive manual handling of reagents and are time-consuming, typically requiring a month or more to complete a single experimental run. Robotic automation of the SELEX procedure can alleviate these issues, but the cost is prohibitive. 24
Microfluidic technology has been emerging to enable rapid isolation of high-affinity aptamers with reduced reagent consumption and in much fewer rounds of iteration.25,26 Microfluidic affinity selection has been performed, in conjunction with off-chip amplification, against targets retained using silica capillary walls, 27 microbeads,28–32 or sol-gels,33–35 or against cells in solution,36,37 to increase selection stringency,28–32,36,37 create more favorable biomolecular environments,32–35 or allow simultaneous positive and negative selections. 38 Moreover, microchips have been developed in an attempt to integrate the overall SELEX process using pneumatically based flow control for isolation of aptamers against viruses, 39 cells,31,40,41 and proteins.42,43 While capable of performing individual affinity selection and PCR amplification processes on chip, these devices apparently require an off-chip procedure to retrieve single-stranded DNA from amplified products between successive rounds of SELEX and do not yet allow full integration of SELEX on a microchip. We have recently demonstrated integration of the entire SELEX process using solid-phase capture and amplification of oligonucleotides on bead surfaces, which are manipulated using either pressure-driven flow or electrokinetics.44,45 In these works, we found that the use of electrokinetic manipulation simplified the device design but involved electric fields that can potentially compromise the integrity of the target or adversely affect the target-aptamer binding. Pressure-driven flow, on the other hand, in general requires extensive flow control operations that complicate the device design and operation.
This article presents a microfluidic approach to full integration of the SELEX process on a microchip. The approach combines the pressure-driven and electrokinetic transport of target-binding oligonucleotides to exploit the advantages of both methods while minimizing their individual limitations. In this hybrid oligonucleotide manipulation method, affinity-selected oligonucleotides are electrokinetically transferred for PCR amplification, while the amplified oligonucleotides are subsequently transferred back using pressure-driven flow for further affinity selection. The hybrid approach simplifies the device design and operation procedures by reduced pressure-driven flow control requirements and avoids the exposure of targets to electric fields prior to and during affinity selection. In addition, bead-based reactions are used to achieve the on-chip coupling of affinity selection and amplification of target-binding oligonucleotides, thereby realizing on-chip loop closure and integration of the entire SELEX process while leveraging the advantages of pressure-driven and electrokinetic transport without requiring offline procedures. The utility of this hybrid approach is demonstrated by isolation of high-affinity aptamer candidates (KD = 12 nM) against the protein immunoglobulin E (IgE) via four rounds of selection and amplification within 10 h.
Experimental
Microbead-Based SELEX Principle
A microbead-based SELEX procedure is integrated on a single chip using combined electrokinetic and pressure-driven transport of oligonucleotides for isolation of aptamers against a given target molecule ( Fig. 1 ). The procedure involves iterative rounds of affinity selection and amplification of target-binding oligonucleotides. Affinity selection occurs on microbeads that are functionalized with a biomolecular target. A randomized library of single-stranded DNA (ssDNA) oligonucleotides, each consisting of a randomized region flanked by two constant primer regions, is then introduced to the beads and binds to the immobilized target. Weakly binding oligonucleotides are removed from the beads by multiple buffer washes. The remaining strongly binding oligonucleotides are released from the beads by thermally disrupting their binding with the target and transferred via electrokinetically migration to a second set of microbeads that are functionalized with reverse primers complementary to the 3′ end of the ssDNA library. The target-binding oligonucleotides are captured by the beads via hybridization to the reverse primers. PCR reagents are then introduced and PCR is performed, resulting in double-stranded DNA (dsDNA) consisting of duplicate copies of (i.e., amplified) target-binding oligonucleotides hybridized to bead-immobilized complementary strands. The amplified target-binding oligonucleotides are released from the beads via thermal denaturation and then transferred via pressure-driven flow to a replenished set of target-functionalized microbeads, on which a new round of affinity selection is carried out without the presence of potentially damaging electric fields. This process is repeated in multiple rounds of affinity selection and amplification to yield aptamers with sufficient affinity to the target.
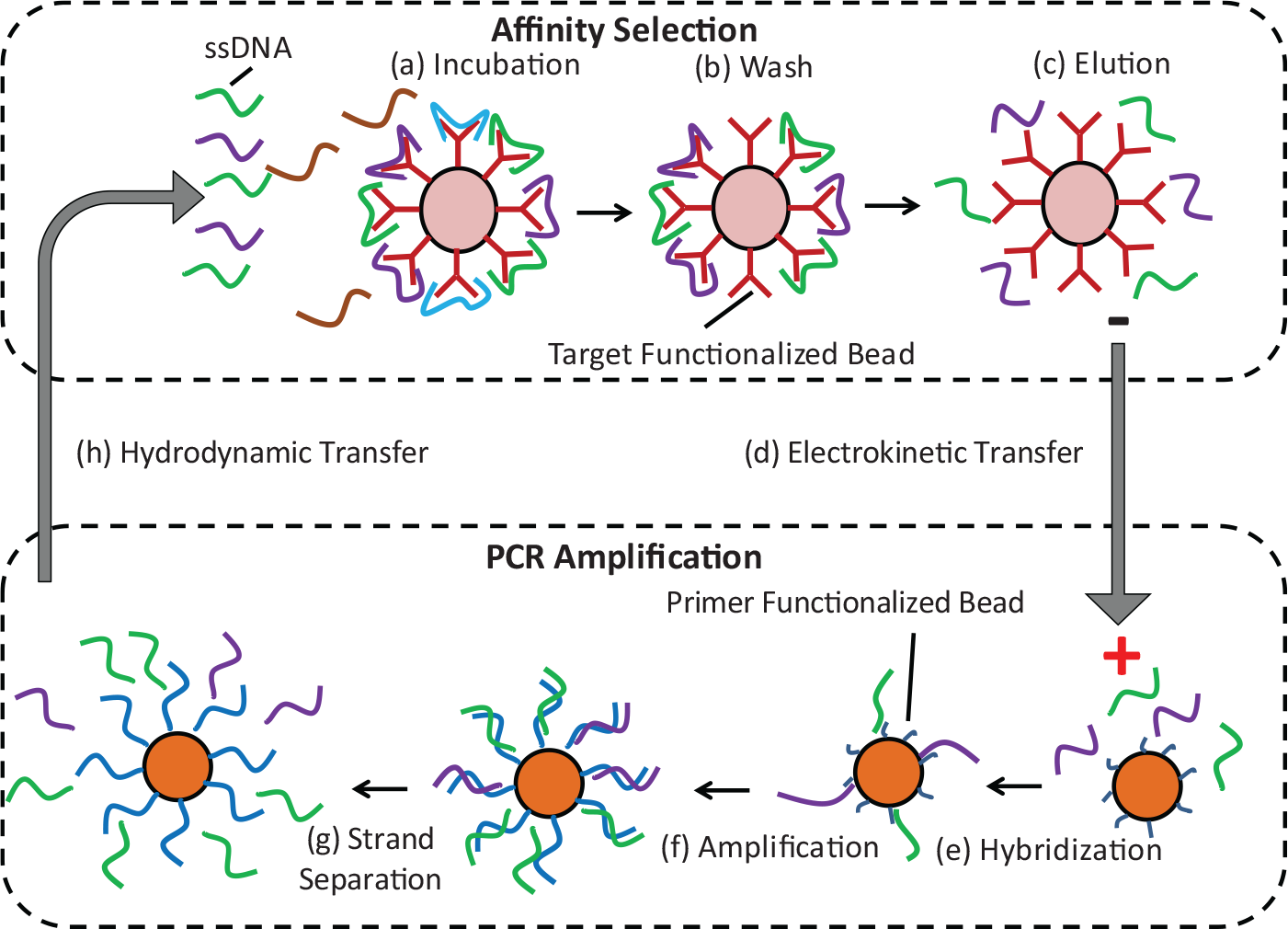
Microbead-based microfluidic systematic evolution of ligands by exponential enrichment (SELEX) principle. (
Microfluidic Device Design and Operation
The microbead-based SELEX procedure is realized in an integrated microfluidic device ( Fig. 2 ). The device consists of two microchambers (respectively referred to as selection and amplification chambers), which are each integrated with thin-film resistive heaters and temperature sensors, and connected to an inlet and outlet used for the introduction of microbeads and buffers, as well as to an electrode port. The selection chamber also includes a microweir, a flow restriction structure (20 µm high) that retains microbeads while allowing fluid passage. The chambers (each 0.5 µL in volume) are interconnected by reagent transport channels. One reagent transport channel (800 µm wide, 240 µm high, and 7 mm long) between the chambers is filled with agarose gel that allows electrokinetically driven oligonucleotide migration while preventing bulk flow ( Fig. 2b ). The other transport channel (600 µm wide, 20 µm high, and 10 mm long), with a semi-circular cross section, lies below a second layer of control channels that are filled with pressurized oil ( Fig. 2c ). Passage of reagent solution in the transport channel is prevented or enabled via changing the oil pressure in the control channels, which hence function as a microvalve. Two supplementary outlet channels sandwich the gel-filled transport channel and are used to allow buffer to fully fill the non-gel-occupied sections of the electrokinetic transport channel. Additional information on the detailed dimensions is included in the Supplementary Materials.
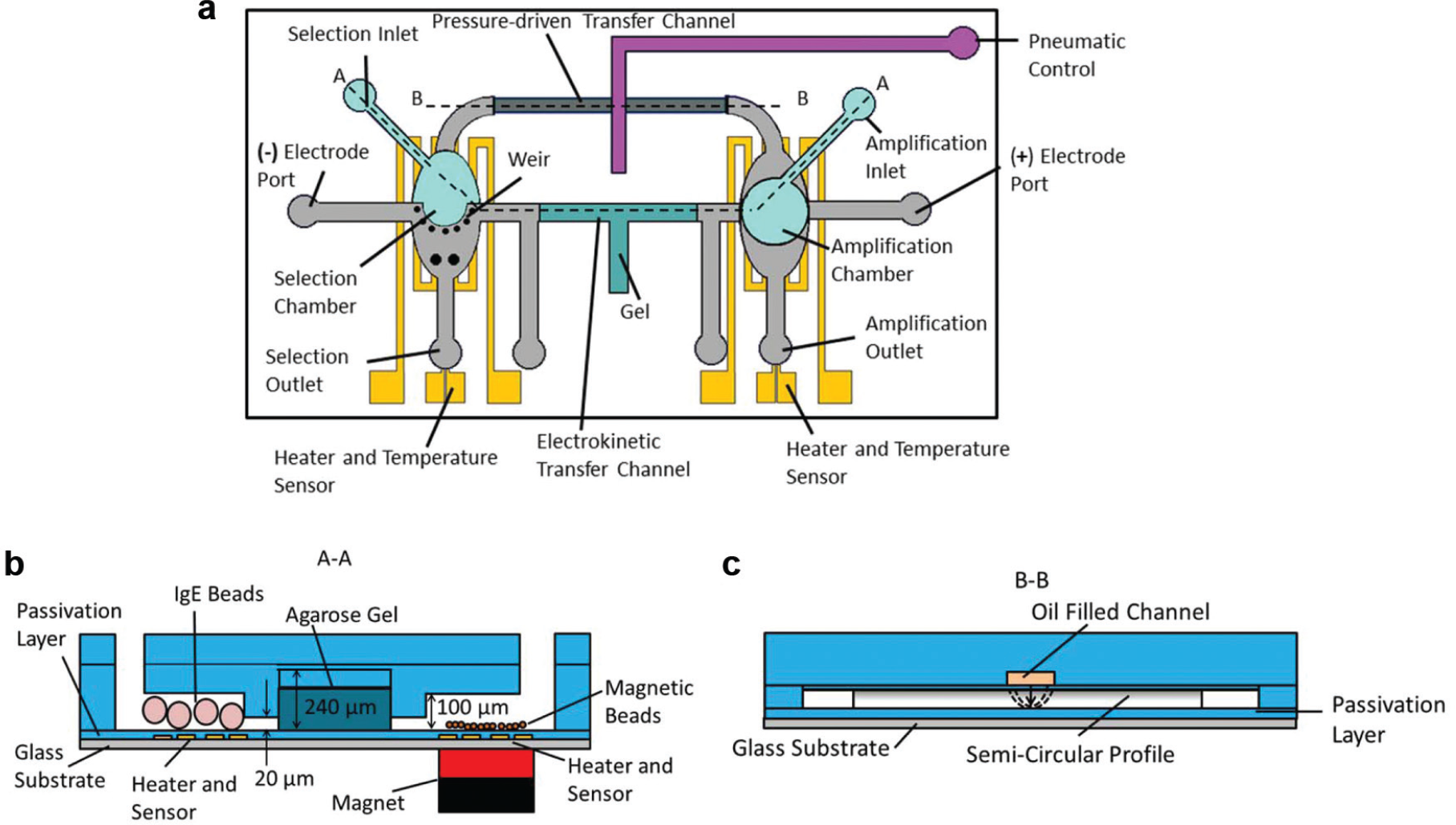
Schematic of the microfluidic systematic evolution of ligands by exponential enrichment (SELEX) device: (
The microfluidic device is capable of repeatedly performing iterative rounds of affinity selection and amplification. Affinity selection occurs in the selection chamber, where target-functionalized microbeads are introduced and immobilized by the microweir structure. A randomized ssDNA library is then introduced to the beads. Strongly binding oligonucleotides are captured by the target, and weakly binding strands are removed by buffer washes. Next, reverse primer-functionalized magnetic beads are introduced into the amplification chamber and held via an external magnet. The oligonucleotides bound to the bead-immobilized target are thermally released from with the selection chamber’s integrated heater, electrokinetically transferred through the gel-filled channel to the amplification chamber, and captured therein by the bead-bound reverse primer. 44 PCR reagents are then introduced and PCR performed using the amplification chamber’s integrated heater and temperature sensor. The amplified ssDNA is thermally released from the beads and, upon the opening of the microvalve, transferred via pressure-driven flow to the selection chamber for further affinity selection. As such, the bead-based protocol enables on-chip separation of amplified ssDNA from complementary strands and on-chip coupling of successive rounds of SELEX, thereby allowing integrated isolation of aptamers.
Materials
Agarose, phosphate-buffered saline (PBS) buffer, MgCl2, Tris, boric acid, and molecular biology grade water were purchased from Sigma-Aldrich (St. Louis, MO). Deoxyribonucleotide triphosphates (dNTPs) and GoTaq Flexi DNA polymerase were obtained from Promega (Madison, WI). Randomized oligonucleotide library (5′–GCC TGT TGT GAG CCT CCT GTC GAA–45N–TTG AGC GTT TAT TCT TGT CTC CC–3′) and primers (forward primer: 5′–FAM–GCC TGT TGT GAG CCT CCT GTC GAA–3′; reverse primer: 5′–dual biotin–GG GAG ACA AGA ATA AAC GCT CAA–3′) were synthesized and purified by Integrated DNA Technologies (Coralville, IA). Human myeloma IgE was purchased from Athens Research and Technology (Athens, GA), and N-hydroxy-succinimide (NHS)-activated microbeads were purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Dulbecco’s PBS (D-PBS) and streptavidin-coupled magnetic beads (Dynabeads M-270 Streptavidin) were purchased from Invitrogen (Carlsbad, CA). AZ-4620 positive photoresist was obtained from Clariant (Somerville, NJ), and SU-8 (2000, 2025, and 2075 series) negative photoresist was purchased from Microchem (Westborough, MA). Polydimethylsiloxane (PDMS) was obtained from Robert McKeown Company (Somerville, NJ), and silicon wafers were purchased from Silicon Quest International (San Jose, CA).
Agarose microbeads functionalized with a protein were used for selection, and magnetic microbeads functionalized with reverse primers were used for amplification. The beads for selection were functionalized with a protein through covalent coupling between the NHS groups of the microbeads and the amine groups of the protein. Prior to an experiment, the protein was incubated with IgE under gentle rotation for 1 h at room temperature and washed with D-PBS buffer. Unreacted NHS groups were passivated by incubating the functionalized beads in Tris buffer. The functionalized beads were then suspended in D-PBS buffer. Similarly, streptavidin magnetic beads were functionalized by incubation with dual biotin-conjugated reverse primers through covalent coupling for 30 min, washed with D-PBS buffer, and suspended in D-PBS buffer.
Microfluidic Device Fabrication
The approach was demonstrated with a PDMS microfluidic device fabricated using standard multilayer soft-lithography techniques. First, a layer of AZ-4620–positive photoresist was spin-coated on a silicon wafer and developed to define the reagent transport channel. Then, using the same silicon wafer, SU-8 layers were spin-coated and developed to define the electrokinetic transfer channel and selection and amplification chambers (collectively termed flow layer). In parallel, SU-8 photoresist was patterned on another silicon wafer to define the layer (termed control layer) bearing the pneumatic control channel. Meanwhile, chrome (10 nm) and gold (100 nm) thin films were deposited and patterned on a glass slide to form heaters and temperature sensors, which were passivated by spin-coating PDMS prepolymer solution and curing at 72 °C for 30 min. PDMS was spun onto the flow layer and poured over the control layer, cured, and peeled off. An inlet hole was punched into the resulting PDMS control layer slab, which was then bonded to the PDMS covering the surface of the flow layer. The resulting multilayer PDMS slab was peeled off the wafer, had inlet and outlets punched, and bonded to the glass slide bearing the heaters and temperature sensors. Finally, molten agarose gel was injected in the gel inlet of the device and allowed to cure at room temperature to form the electrokinetic transfer channel, and mineral oil was injected into the pneumatic control channel. A fabricated device is shown in Figure 3 .

Photograph of a fabricated microfluidic device filled with dye solutions for visualizations. Scale bar: 5 mm. PDMS, polydimethylsiloxane.
Measurement of Electrokinetic Effects on Chamber Properties
Chamber pH was observed to evaluate the potential deleterious effects of prolonged electric fields on the selection chamber. A fabricated device was filled with TB buffer, and an electric field (25 V/cm) was applied for varying lengths of time (0–30 min) with cathode located in the selection chamber and anode located in the amplification chamber. After the prescribed amount of time elapsed, the chamber volume was collected and the pH was measured. The process was repeated three times for each time course.
Integrated Aptamer Selection Procedure
The fabricated device was connected to a syringe pump (New Era Pump Systems, Farmingdale, NY), a multimeter (34420A; Agilent Technologies, La Jolla, CA), a power supply (E3631; Agilent Technologies), and a nitrogen tank with pressure regulator (Concoa, Virginia Beach, VA). The syringe pump was used to introduce buffers, reagents, and beads. The multimeter and power supply measured the resistance of the temperature sensor and supplied power to the heater, respectively, and were connected to a computer that implements a LabVIEW code for closed-loop temperature control of the device. The pressure regulator was connected to the pneumatic control layer and allowed the oil-filled channel to be pressurized, thereby closing the valve.
Integrated SELEX occurred on chip without the use of any offline processes. Protein functionalized microbeads were injected into the selection chamber of the device (20 µL/min) until approximately 40% of the selection chamber volume was occupied by beads. Selection of oligonucleotides was then performed by infusing a randomized library (1 µM in D-PBS buffer) into the device (10 µL/min) for 10 min, followed by multiple washes with D-PBS buffer (20 µL/min) to remove weakly binding oligonucleotides, which were collected in a tube and stored. Collected oligonucleotides were amplified off chip (16 rounds of PCR) and imaged with gel electrophoresis to verify successful removal of weakly binding oligonucleotides and retention of strongly binding oligonucleotides that bind to targets functionalized on bead surfaces.
Next, primer-functionalized magnetic beads were introduced into the amplification chamber of the device and held by an external magnet located beneath the chamber. Tris–boric acid electrolyte buffer (89 mM Tris, 89 mM boric acid, and 100 mM NaCl) was then injected into the device and platinum wires were inserted into the electrode inlets of each chamber to generate an electric field. Meanwhile, strongly bound oligonucleotides remaining in the selection chamber were thermally released (50 °C) using the integrated heater and temperature sensor. The released oligonucleotides were transferred from the selection chamber to the amplification by applying a 25-V/cm electric field for 40 min.
Following transfer of the oligonucleotides to the amplification chamber, the electric field was removed. PCR reagents were then introduced into the amplification chamber and bead-based PCR progressed using the heater and temperature sensor located beneath the amplification chamber. A PCR process of 95 °C for 10 s, 59 °C for 30 s, and 72 °C for 10 s was used. After 20 cycles of PCR thermocycling, the protein-functionalized microbeads were removed from the selection chamber and replaced with new protein-functionalized microbeads. Bead-based PCR was confirmed by introducing a fluorescently modified oligonucleotide library (100 pM) into the amplification chamber containing reverse primer–functionalized beads and performing 20 cycles of PCR thermal cycling. Bead surfaces were imaged using an epifluorescence microscope (IX 71; Olympus, Center Valley, PA) equipped with a CCD (c8484; Hamamatsu, Boston, MA) before and after PCR, as well as after thermally induced ssDNA release. Additional information on the detailed procedure is included in the Supplementary Materials.
Following amplification, the valve was then opened and oligonucleotides were released from the bead surfaces by heating to 95 °C. The released oligonucleotides were transported back to the selection chamber through the opened valve via pressure-driven flow (20 µL/min) for further affinity selection with the replenished microbeads. This closed-loop process was repeated for a total of four affinity selections and three 20-cycle PCR amplifications.
Aptamer Characterization Procedure
The enriched aptamer pool collected following completion of SELEX was investigated for its affinity and specificity using a fluorescence binding assay. 1 Six different concentrations (100 nM, 50 nM, 25 nM, 12.5 nM, 6.25 nM, and 3.125 nM) of fluorescently tagged oligonucleotides were incubated with IgE-functionalized beads in triplicate 100-µL volumes. After incubating the oligonucleotides with the beads for 30 min, the beads were washed and the bound oligonucleotides were thermally eluted (95 °C). The eluted oligonucleotides were collected and their relative amounts were determined with a Wallac EnVision Multilabel Reader (PerkinElmer, Waltham, MA) fluorescent spectrometer. The relative amounts were analyzed with a hyperbola saturation binding curve fitting (1:1 binding) using GraphPad Prism (GraphPad Software, La Jolla, CA) to determine the binding affinity of the oligonucleotide pool toward the target molecule. The specificity of the aptamer candidate pool toward IgE was investigated by repeating the binding study with bare beads and beads that were functionalized with immunoglobulin A (IgA).
Results and Discussion
We first present results from the characterization of the individual components of the microfluidic device, including pH effects, affinity selection, PCR amplification, and loop closure. We then describe results from the fully integrated SELEX process as well as the characterization of the resulting aptamer candidates. The protein IgE was used as a representative molecule throughout the experiments.
Electrokinetic Effects on Chamber pH
To demonstrate the benefits of the hybrid approach’s ability to avoid the potentially deleterious effects of using each method alone, we characterized the electrokinetic transfer process. The selection chamber pH was evaluated over various time courses ( Fig. 4 ) with electrodes oriented to allow migration from the amplification chamber to the selection chamber. A significant pH drop from 8.3 to 7.25 was observed in the selection chamber after the application of an electric field for 30 min, which can be attributed to the generation hydroxide (OH–) and hydrogen (H+) ions at the cathode and anode, respectively.46,47 Thus, the electrolysis of buffer during electrokinetic transport of DNA can induce changes in the pH level microchambers, which can potentially compromise the integrity of the target and disrupt the electrostatic interactions between the aptamer and oligonucleotides.46,47 While electrokinetic transport can significantly affect affinity selection, the hybridization interactions between complementary oligonucleotides are more robust, and for pH 5–9, the renaturation rate is practically independent of pH. 48 Thus, while electrophoresis of oligonucleotides from the amplification chamber to the selection chamber may not be desirable, electrokinetic oligonucleotide transport from the selection chamber to the amplification chamber is appropriate. These observations are consistent with the rationale for our hybrid oligonucleotide transport approach.
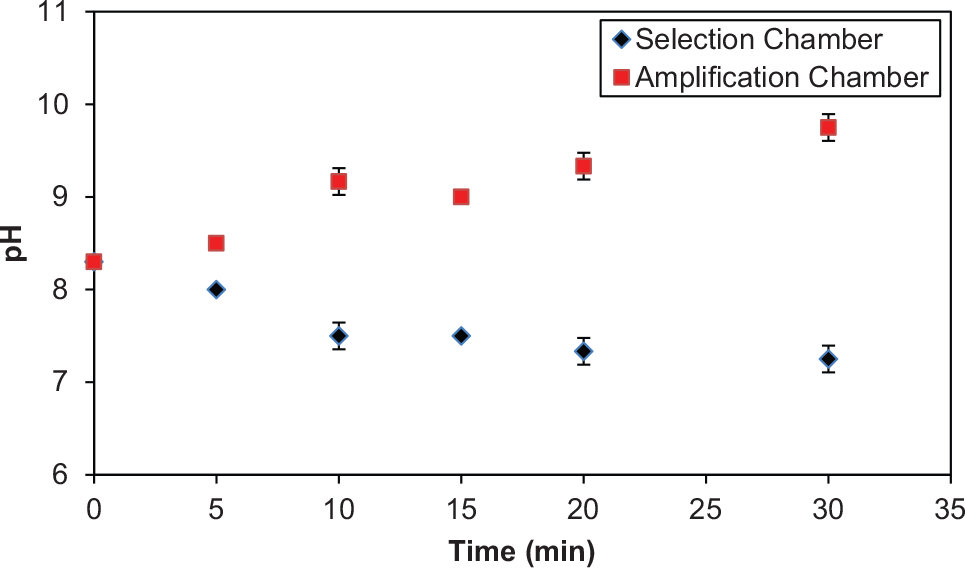
Electrokinetic effects on chamber pH. Electric field was applied for different time lengths (0–30 min), and the chamber pH was measured. Error bars represent standard deviations from triplicate measurements.
Characterization of Affinity Selection
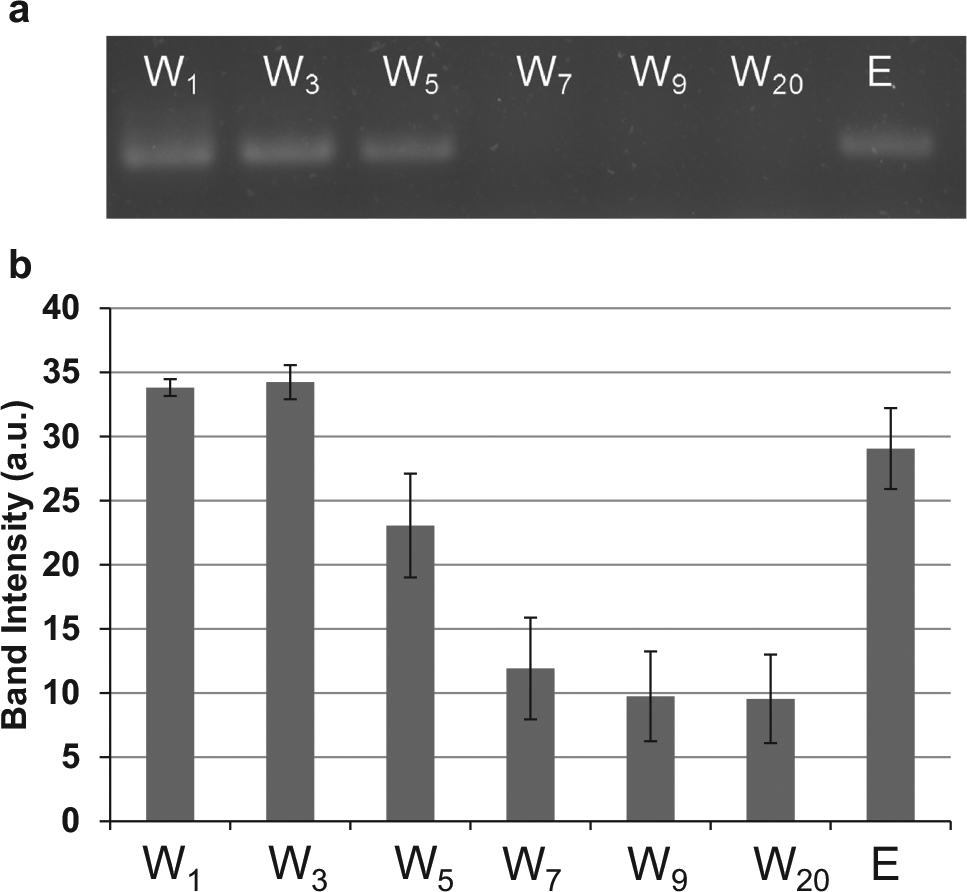
Eluates from affinity selection were collected and evaluated using gel electrophoresis (
Fig. 5
). A band in the electropherogram indicates the presence of oligonucleotides; thus, the strong band in the first wash (W1) demonstrates that not all oligonucleotides bound to the target molecule and some were flushed out of the device during the washing stage. The decrease in band intensity from wash 1 to wash 20 shows that as washing progressed, fewer oligonucleotides were eluted from the target, while the strong band intensity in the elution lane (E) demonstrates that oligonucleotides strongly bound to the bead surfaces were eluted when heating the chamber to 50 °C. Therefore, the selection process partitioned nonbinding oligonucleotides from binding oligonucleotides, which could then be eluted by increasing the temperature. The thermally eluted oligonucleotides can be electrokinetically transferred (
(
Characterization of Bead-Based PCR Amplification and Loop Closure
To characterize the bead-based PCR protocol, randomized library strands were amplified on chip using a fluorescently tagged forward primer and monitored with fluorescent microscopy (
Fig. 6
). During the amplification process, dsDNA was generated consisting of fluorescently labeled amplified library strands hybridized to their bead-bound complements. Thus, the increase in fluorescent intensity of beads following 20 cycles of PCR indicates that template oligonucleotides were successfully amplified on the bead surfaces within the device. The subsequent decrease in fluorescent intensity of the bead surfaces upon heating to 95 °C demonstrates that the fluorescently modified strands dehybridized from the complementary biotinylated strand bound to the bead surface. Affinity-selected oligonucleotides can be transferred via electrokinetics (details in Supplementary Material) to bead-based PCR to allow closed-loop aptamer enrichment. Eluates collected during the closed-loop process (
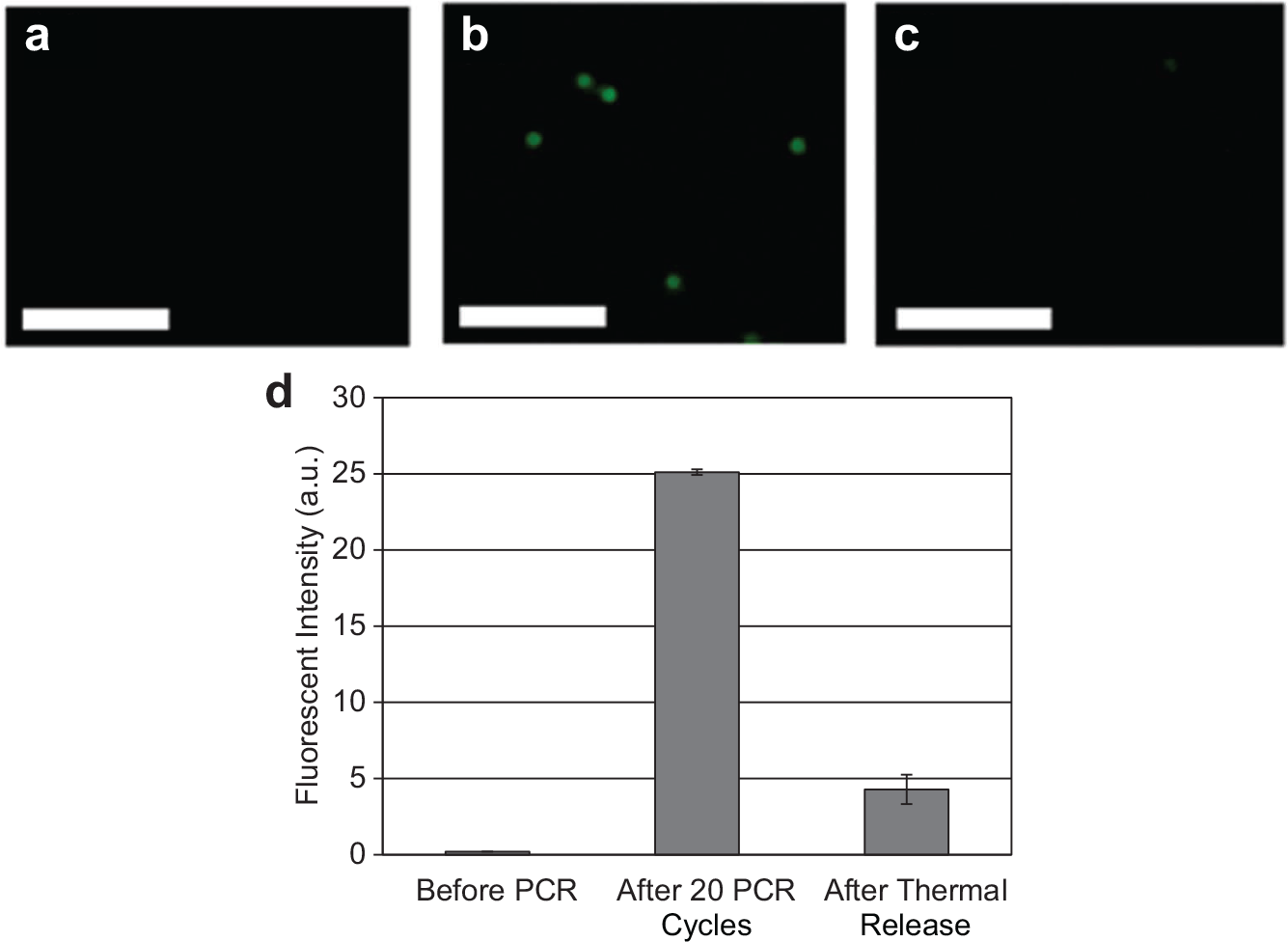
Fluorescent images of beads (
Integrated Multiround SELEX for Isolation of Aptamers
Having confirmed the approach’s potential for closed-loop affinity selection and amplification, a microchip was used to demonstrate multiround hybrid SELEX where four rounds of affinity selection and three rounds of PCR amplification were performed on chip ( Fig. 7 ). Eluates were collected during each round of affinity selection, amplified by PCR, and analyzed using conventional gel electrophoresis. Since the brightness of bands in a gel image represents the amount of oligonucleotides in the eluent loaded in the lane, comparison of the band intensities allowed investigation of the SELEX process. In the first round, some oligonucleotides were in the washing waste after the completion of the washing process, as indicated by the presence of a band in lane S1W9. However, the increase in band intensity from S1W9 (selection 1, wash 9) to S2W1 (selection 2, wash 1) suggests that oligonucleotides were released from the bead surfaces, electrokinetically transferred, and amplified by PCR. The lack of a band in S2W9 (selection 2, wash 9) shows that weakly bound oligonucleotides were removed from the washing process. The increase in band intensity in from S2W9 to S3W1 (selection 3, wash 1) and S3W9 (section 3, wash 9) to S4W1 (selection 4, wash 1) suggests that, again, oligonucleotides were thermally released, electrokinetically transferred to the amplification chamber, amplified by PCR, and transferred back to the selection chamber via pressure-driven flow. The presence of oligonucleotides in the elution lane (E) and the lack of detectable oligonucleotides in S4W9 (selection 4, wash 9) suggest that oligonucleotides were strongly bound to the target after four rounds of selection and amplification and were successfully released from the bead surfaces.
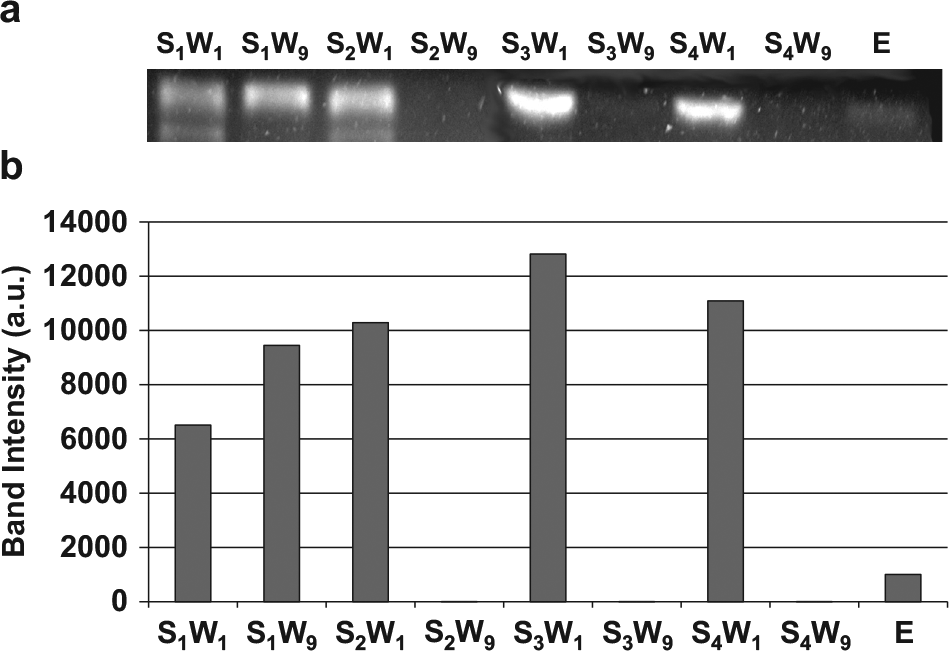
(
Binding Affinity and Specificity of Aptamers
The resulting aptamer candidate pool was analyzed for its affinity and specificity toward its intended target molecule. When the enriched pool was incubated with IgE-immobilized beads, washed, and bound oligonucleotides were thermally eluted and measured, the fluorescent intensity rapidly increased until reaching an asymptote ( Fig. 8 ). This indicated that the affinity of the enriched oligonucleotide pool considerably improved after the microfluidic SELEX process. Because this measurement relied on many manual transfers, the error increases with higher concentrations of binding aptamer candidates. The data were well represented by a Langmuir monovalent binding model, with an equilibrium dissociation constant (KD) of approximately 12 nM (obtained via nonlinear regression), as is consistent with that of existing IgE aptamers.6,44 In contrast, when the enriched pool was incubated with IgA-functionalized beads or bare beads, washed, and bound oligonucleotides were eluted and measured, the fluorescently increased very slowly with the oligonucleotide concentration. This demonstrates the aptamer candidate pool binds much more strongly to the IgE-functionalized beads than the bare beads, indicating the aptamer is indeed specifically binding to the protein target.
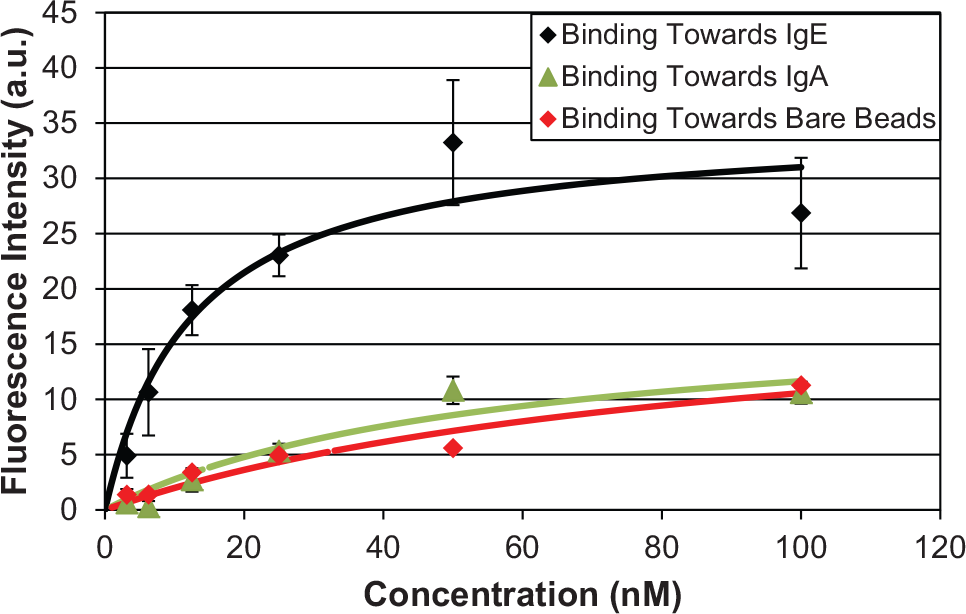
Fluorescence-based binding affinity measurements of enriched pool toward immunoglobulin E (IgE) functionalized, immunoglobulin A (IgA) functionalized, and bare microbeads. Error bars represent standard deviations from triplicate measurements.
In conclusion, a microfluidic approach to integrated isolation of aptamers against biological targets has been described. The approach employs a bead-based protocol for affinity selection and amplification of oligonucleotides, which are transported within the protocol via combined electrokinetic transport and pressure-driven flow. Specifically, oligonucleotides are affinity selected against target-functionalized beads and binding strands are transferred under an electric field for bead-based PCR amplification. The amplified product is then transferred back for further affinity selection through pressure-driven flow without relying on off-chip processes. This hybrid approach simplifies the microfluidic device design and operation by reduced pressure-driven flow control requirements and avoids potentially deleterious effects of the exposure of targets to electric fields prior to and during affinity selection. The approach thus allows the entire iterative SELEX process to be integrated on a single chip without the requirement for any offline procedures, thereby enabling rapid isolation of aptamers.
The integrated hybrid microfluidic SELEX approach was demonstrated by isolating aptamers with high affinity toward the protein IgE. Binding oligonucleotides are affinity selected against IgE-functionalized beads, electrokinetically transferred to an amplification chamber, hybridized to microbeads, amplified by PCR, and transferred back to the selection chamber by pressure-driven flow for a new round of affinity selection. The SELEX process was completed in four rounds within a total runtime of approximately 10 h. The resulting aptamer candidates displayed strong target-binding affinity, with an equilibrium dissociation constant of 12 nM. These results demonstrate the utility of the approach to rapidly isolate aptamers with high affinity to biological targets.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support from the National Science Foundation (Award Nos. CBET-0854030, CBET-1033288, and CBET-1026591), the National Institutes of Health (Award Nos. 8R21 GM104204, R01GM104960, 1R21CA199849, UL1 TR000040, and TL1 TR000082), and the Chinese Academy of Sciences SAFEA International Innovation Teams program.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.