
Abstract
High-throughput, direct measurement of substrate-to-product conversion by label-free detection, without the need for engineered substrates or secondary assays, could be considered the “holy grail” of drug discovery screening. Mass spectrometry (MS) has the potential to be part of this ultimate screening solution, but is constrained by the limitations of existing MS sample introduction modes that cannot meet the throughput requirements of high-throughput screening (HTS). Here we report data from a prototype system (Echo-MS) that uses acoustic droplet ejection (ADE) to transfer femtoliter-scale droplets in a rapid, precise, and accurate fashion directly into the MS. The acoustic source can load samples into the MS from a microtiter plate at a rate of up to three samples per second. The resulting MS signal displays a very sharp attack profile and ions are detected within 50 ms of activation of the acoustic transducer. Additionally, we show that the system is capable of generating multiply charged ion species from simple peptides and large proteins. The combination of high speed and low sample volume has significant potential within not only drug discovery, but also other areas of the industry.
Keywords
Introduction
The use of mass spectrometry (MS) in the biological and chemical sciences has grown exponentially over the last two decades. The introduction of commercially available mass spectrometers fitted with electrospray sources in the late 1980s and early 1990s enabled the analysis of a wide range of molecular species, from druglike small molecules to large proteins and cellular metabolites. The ability to couple electrospray to separation techniques, such as high-performance liquid chromatography (HPLC), meant that complex mixtures could be separated and characterized in one process. The ionization mechanism of electrospray generates mainly protonated or deprotonated ions, depending on the ionization mode. This produces spectra that generally contain just molecular ions with very little fragmentation, thus simplifying interpretation of datasets. 1 The technology has been applied across a wide range of disciplines and has enabled advances in areas such as drug metabolism, 2 proteomics, 3 metabolomics,4,5 lipidomics, 6 and imaging. 7 The increasing ease of use and robustness of these once complex systems, combined with the provision of myriad software tools to confidently extract information from data, have moved this instrumentation into the hands of the bench scientist.
While the application areas for mass spectrometry have increased, its basic throughput has not changed significantly over the same time frame. This is predominantly due to the requirement that samples be subjected to a separation technique prior to introduction to the mass spectrometer to isolate analytes of interest from signal-suppressing matrix components. Improvements in HPLC system design, such as reductions in dead volumes and an increase in pumping pressure, have enabled the benefits of smaller columns containing smaller particles, improved separation, and faster run time to be realized. Despite these improvements, the time required for simple separation is still around 1 min. 8 Even if real separation is not required, the mechanics of getting samples into the mass spectrometer still limit sample loading time to about 10 s per sample.
There has been some success in improving throughput performance by overcoming matrix effects and increasing analysis speed. Simplifying sample processing by using solid-phase extraction, rather than traditional chromatography, to remove salts can reduce preinjection times to under 10 s per sample from the minutes per sample required for HPLC. 9 However, the increase in sampling speed comes at the cost of sensitivity. Processing samples in parallel using multiplexed chromatography systems can increase throughput, 10 but the complexity of this approach can negatively impact system reliability and often preclude its use in HTS environments.
Techniques that rely on a laser to deliver ionization energy and free analytes from the sample matrix also offer some improvements in speed. In matrix-assisted laser desorption ionization (MALDI), ionization of the sample is a secondary process where laser energy is absorbed by either nanostructures in the plate surface topography or a matrix molecule,11,12 usually substituted cinnaminic or picolinic acids. These excited molecules in turn ionize the target molecule via charge transfer. MALDI works well for peptides, small proteins, lipids, and oligonucleotides13–15 and can be performed at speeds of 1 s per sample, but underperforms for a wide range of small molecules. 16 A related technique, laser diode thermal desorption (LDTD), desorbs sample directly into the gas phase via a thermal pathway. However, application of LDTD in the literature has been mainly aimed at small druglike molecules, as the thermal nature of this technique and the use of an ambient pressure chemical ionization (APCI) system make it unsuitable for both modified peptides and cellular metabolites in biochemical screening. Additionally, LDTD is slower than MALDI, requiring around 10 s per sample. 17
The work presented here demonstrates the possibility of using acoustic droplet ejection (ADE) to deliver sample directly from individual microtiter plate wells to a mass spectrometer. An ultrasonic pulse is delivered to the well of interest, ejecting a “mist” of femtoliter-sized droplets. These droplets are charged via the application of an electric field. The charge is then passed to the analyte as the droplets desolvate in transit to the mass spectrometer. We demonstrate the ability to generate signal from small molecules and multiply charged peptides and proteins, as well as sampling speeds of up to three samples per second.
Materials and Methods
Acoustic loading of a mass spectrometer was accomplished according to the basic schema shown in Figure 1 . While the data presented here were collected over a number of specific iterations and equipment changes to the system, the basic design remained the same. The acoustic transducer assembly of an Echo 555 (Labcyte, Sunnyvale, CA) was set up externally, though it remained connected to the Echo fluidics and electronics via an umbilical cable. While significantly modified to mate to the MS system, the Labcyte Echo used in the Echo-MS retained the full set of features found in other Echo systems. These include Dynamic Fluid Analysis (DFA), which allows real-time measurement and adjustment of the acoustics to maintain ejection over a wide range of fluids, including blood, plasma, cell culture medium, acid digests, and chemical syntheses.

An early Echo-MS prototype that outlines the basic instrument schema, including the externalized Echo transducer bubbler assembly (
Samples were acoustically ejected from microtiter plate wells positioned directly above the transducer. Both 384-well polypropylene (PP) and 384-well cyclic olefin copolymer (COC) plates were used (Labcyte). In later versions of the instrument, a motorized XY stage was added to allow for automated sampling of multiple wells. Acoustic ejection, volume transfer, and plate movement were controlled using the Cherry Pick software program (Labcyte), though the underlying firmware was heavily modified to meet the novel needs of this hybrid Echo-MS instrument prototype. Ejection events were performed at a rate of 500 Hz. Droplets ranged from the Labcyte Echo 555 standard 2.5 nL (167 µm diameter) down to a mist of microdroplets, whose diameters were found to be from microns to tens of microns ( Fig. 2 ). The flow rate from a well was calculated to be around 4 µL/min and was found by monitoring the rate of fall of the liquid meniscus during analysis using the well survey function within Labcyte’s Echo control software. All MS signals produced were a sum of a number of misting events dependent on the scan or dwell time of the experiment performed.
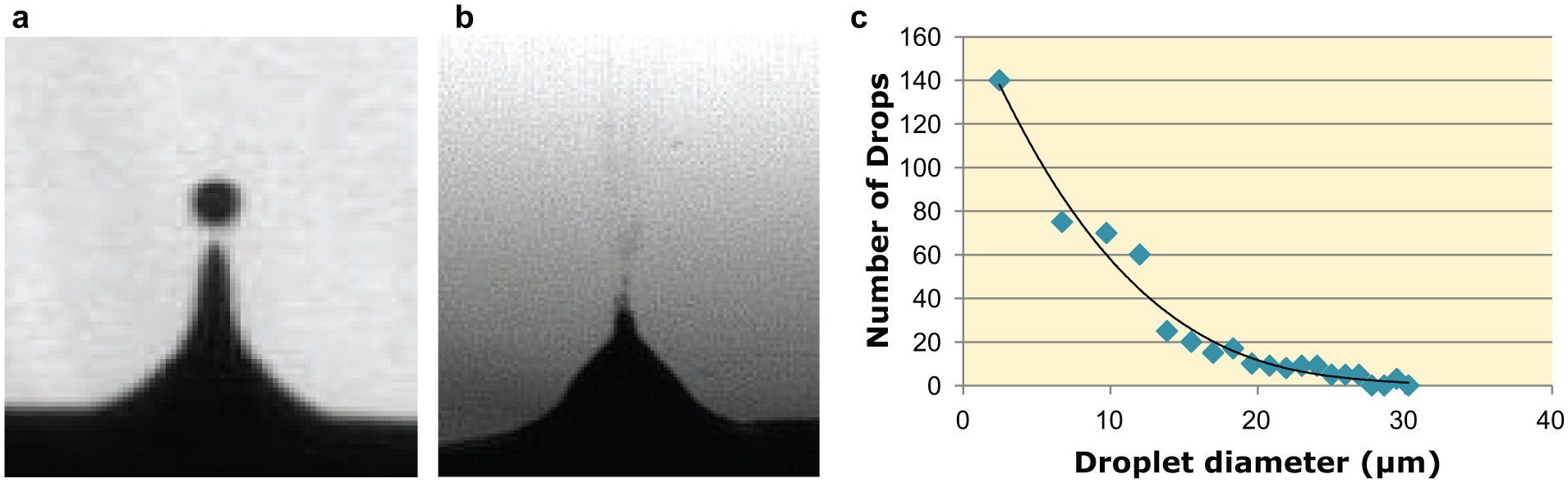
Photographs of acoustically ejected droplets. Samples were first ejected as discrete 2.5 nL (167 µm) droplets as per the standard operation of an Echo 555 (
A transfer tube interface containing a vacuum restrictor was attached to the front end of the MS and allowed for proximal access to the ejected droplets via suction. The transfer tube was heated to 180 °C to promote desolvation of ejected droplets and production of free ionized species. 18 The two mass spectrometers used for this work were a Xevo G2S QTOF and a SQD II single quadrupole (Waters, Wilmslow, UK) and were operated using MassLynx 4.1 software. Unless otherwise specified, the QTOF scanned a mass range of 50–1200 Da in 1 s. The SQDII was operated in single ion recording (SIR) mode monitoring the mass of interest with a 50 ms dwell time. All data apart from those shown in Figures 3 and 9 were generated on the SQD II. Figures 6 and 7 were generated in full-scan continuum mode on the SQD II, scanning from 100 to 500 Da and from 500 to 1600 Da, respectively, both with a scan time of 0.5 s. All units associated with mass spectrometers are arbitrary units (a.u.); even those declared as total ion counts (TICs) are a number based on the range of the analog-to-digital converter for that particular instrument model.
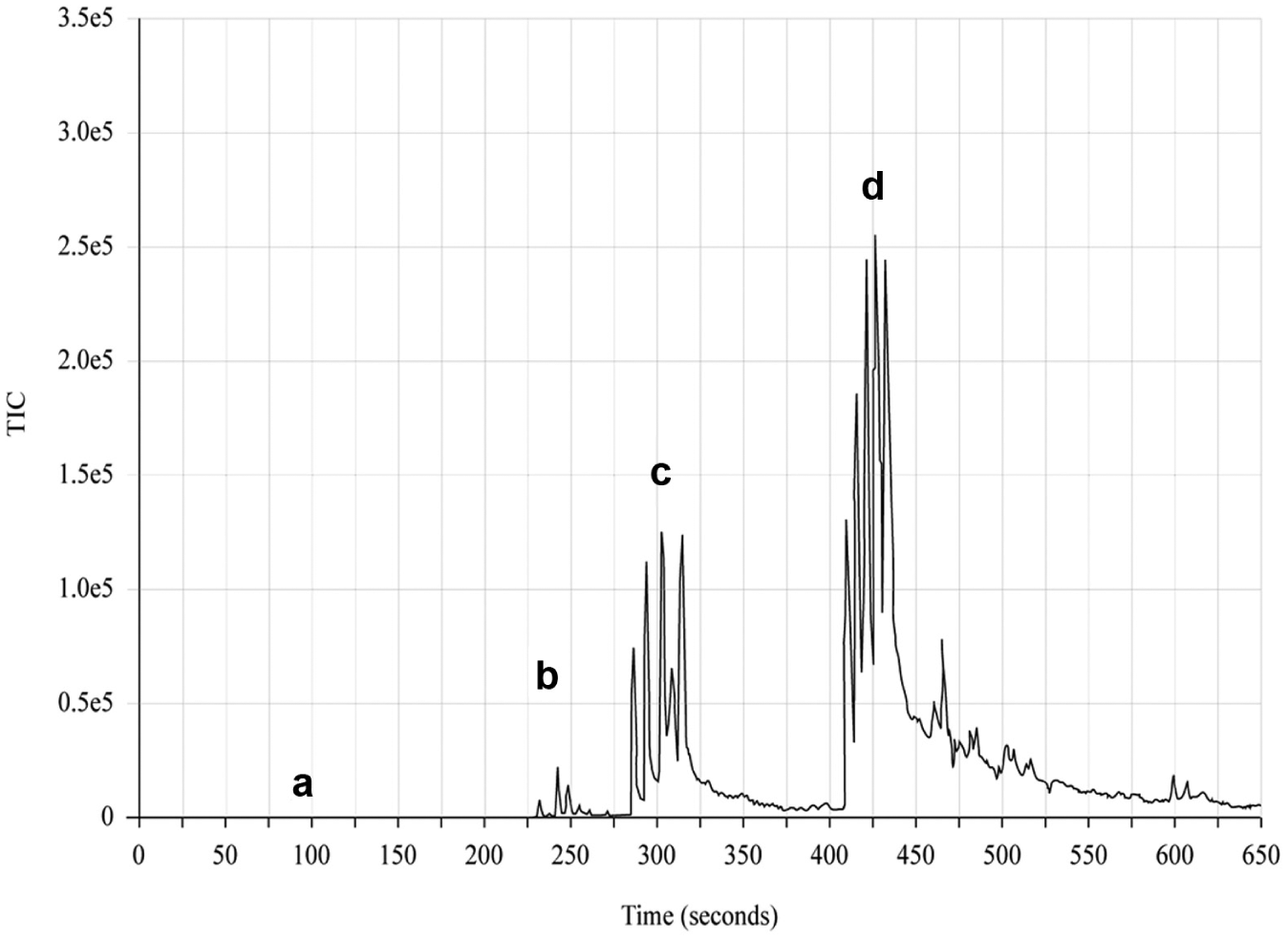
A mass chromatogram, at m/z 311, of various acoustic ejections of sulfadimethoxine. Initially no signal was generated using the standard Echo 555 2.5nL droplet (
The performance of the Echo-MS was gauged via a set of standard analytes: sulfadimethoxine (311 m/z in positive mode), caffeine (195 m/z in positive mode), and warfarin (309 m/z in positive mode, 307 m/z in negative mode), each used at concentrations of 10 and 100 µM. Caffeine can be readily ionized via various atmospheric pressure ionization (API) mechanisms, making it an excellent model system upon which to iterate the design of the Echo-MS (data not shown). Additionally, warfarin has been well characterized at AstraZeneca as a standard quality control (QC) sample for monitoring electrospray source performance on their liquid chromatography–mass spectrometry (LC-MS) platforms. Glu-1-fibrinopeptide B (glu-fib; 1.5 kDa) and ubiquitin (10 kDa) were used to gauge performance of doubly and multiply charged species in the positive mode, respectively. All test samples were obtained from Sigma-Aldrich (Haverhill, UK) and dissolved in pure water (MilliQ), unless otherwise specified.
Initially, analyte-containing charged droplets were produced via acoustic ejection of neutral droplets into a stream of ions produced by a secondary electrospray ionization
19
(SESI) source (
Initial measurements using this drop charging approach involved placement of a wire directly into the well fluid. A high-voltage power supply was connected to the wire and typically set at 4 kV, while the free end of the MS transfer tube was held above the well, and maintained at the cone voltage of the mass spectrometer (magnitude typically <100 V). In a later implementation, a resistive glass capillary (Photonis, Lancaster, PA) was attached to the end of the MS transfer tube. The free end of the glass capillary was then placed above the well fluid and could be held at high voltage, typically 5 kV, while the well fluid itself was allowed to float electrically, with no wire present in the well. The transfer tube on the other end of the resistive glass capillary was again held at the cone voltage of the mass spectrometer. The polarity of the high voltage applied to the free end of the resistive capillary could be automatically switched after a specified amount of time via Echo firmware, to avoid excessive charging of the sample well fluid, as charge was removed over time with the ejected drops.
Results
As a proof of principle to demonstrate ion generation using acoustic droplets, we ejected a sulfadimethoxine aqueous solution through a SESI ion stream into the MS transfer tube interface. The chromatogram presented in Figure 3 shows the output of real-time tuning of the acoustic ejection process while monitoring the mass ion of sulfadimethoxine. During the initial section of the chromatogram, 2.5 nL droplets were generated and produced no signal. Droplet size was sequentially reduced at 230, 300, and 425 s in the time trace via retuning of the acoustic ejection pulse, resulting in increasing signal intensity. The multiple peaks and high background signal produced during this experiment were attributed to suboptimal conditions for both the SESI mixing stage and the MS transfer tube. While the data shown in Figure 3 are of relatively poor quality, they demonstrated early on that droplet size and, by extension, the nature of the acoustic pulse had a significant impact on the potential for droplet ionization in this configuration. Although it was understood that the changes made to the acoustic pulse would lead to a reduction in the average droplet size, at this stage we had yet to characterize the droplet population.
After removal of the SESI from the setup, charged droplets were generated by applying a bias voltage directly to the well fluid via a wire placed in the well, while maintaining the MS transfer tube at the MS cone voltage and positioning the collection nozzle above the well fluid. Under the presence of these field conditions, 60 µM glu-fib solution was acoustically ejected as a mist of charged droplets. Analytes became charged as the droplets desolvated in the heated transfer tube while en route to the front end of the MS. The average ion abundance and standard deviation of thirty 0.3 s ejections are shown in Figure 4a (wire in a well). Here each ejection is the sum of a collection of misting events.
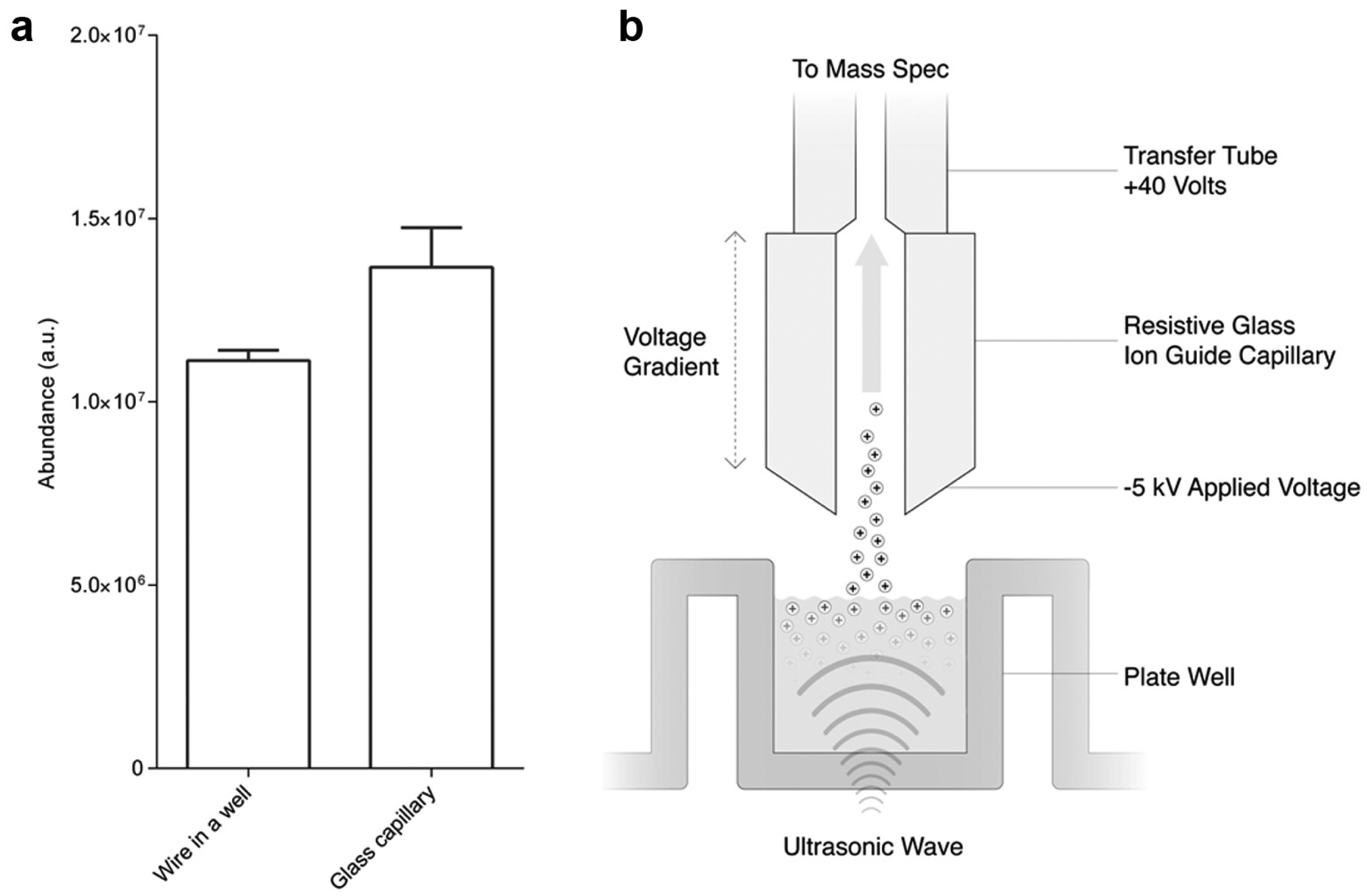
(
To enable whole-microtiter plate analysis, it was necessary to decouple the mechanism of droplet charging from the well and, ultimately, allow measurement across an unmodified plate. To achieve this goal, first a resistive glass capillary was fastened to the end of the transfer tube with its well-facing inlet connected to the high-voltage supply. Application of a voltage potential of the opposing polarity to the ion charge desired generated an electric field between the fluid surface and the capillary inlet, inducing charge on the droplets as they were ejected from the fluid surface ( Fig. 4b ). The resistive nature of the glass capillary then allowed this high voltage to cascade down its length so as to be compatible with the 40–50 V present on the transfer tube. The performance of the glass capillary was confirmed via acoustic ejection of 60 µM glu-fib solution from a well wired to electrical ground where, again, the average ion abundance and standard deviation of thirty 0.3 s ejections are shown ( Fig. 4a , glass capillary). The data produced are qualitatively similar to those of the original transfer tube without the capillary attachment.
The situation became more complicated, however, when the grounding wire was removed from the well and the well was allowed to electrically float. Because the plate material is electrically insulating, significant current could not flow into the wells to replace the charge lost due to droplet ejection. This resulted in a net charge buildup in the sample fluid such that the voltage potential of the liquid eventually reached a value close to the applied extraction potential, reducing the extraction field. Since the extraction field is responsible for inducing charge onto the ejected droplets, at this point no more ions were detected in the mass spectrometer. In general, this phenomenon was witnessed as an exponential decay in time of the MS signal as drops were ejected at a constant rate from the well. The time constant of this decay in the MS signal could be related to the effective capacitance of the well, the current that was ejected from the well via droplets, and the extraction voltage itself.
For typical experimental values of drop ejection rate, volume, and drop charge, the time constant in 384-well PP plates was found to be approximately 3 s. For the lower-volume 384-well COC plates, the time constant was found to be less than 0.5 s. These times were consistent with expectation from simple analysis of the induced drop charge and well capacitance.
To avoid the effects of this decay in the MS signal from the insulated wells, a scheme was employed to periodically switch the polarity of the extraction voltage at the glass capillary while maintaining acoustic ejection. Thus, the charge accumulated in the well fluid over one half cycle of the switched extraction voltage profile ( Fig. 5 ) would be dissipated over the following half cycle. Ideally, the period of polarity switching would be significantly smaller than the decay time of the MS signal, in order to minimize the charging of the well fluid. Figure 5 shows signal generated by 60 µM glu-fib solution acoustically ejected from an electrically floating well using this technique. Total acoustic ejection time is 6 s, and the high-voltage polarity was switched every second. The signal from the well shows the characteristic picket fence pattern due to periodic cycles of charging and discharging the well. Switching voltages at these speeds has little impact on the transit of ions into the mass spectrometer at the transition of this switching event.
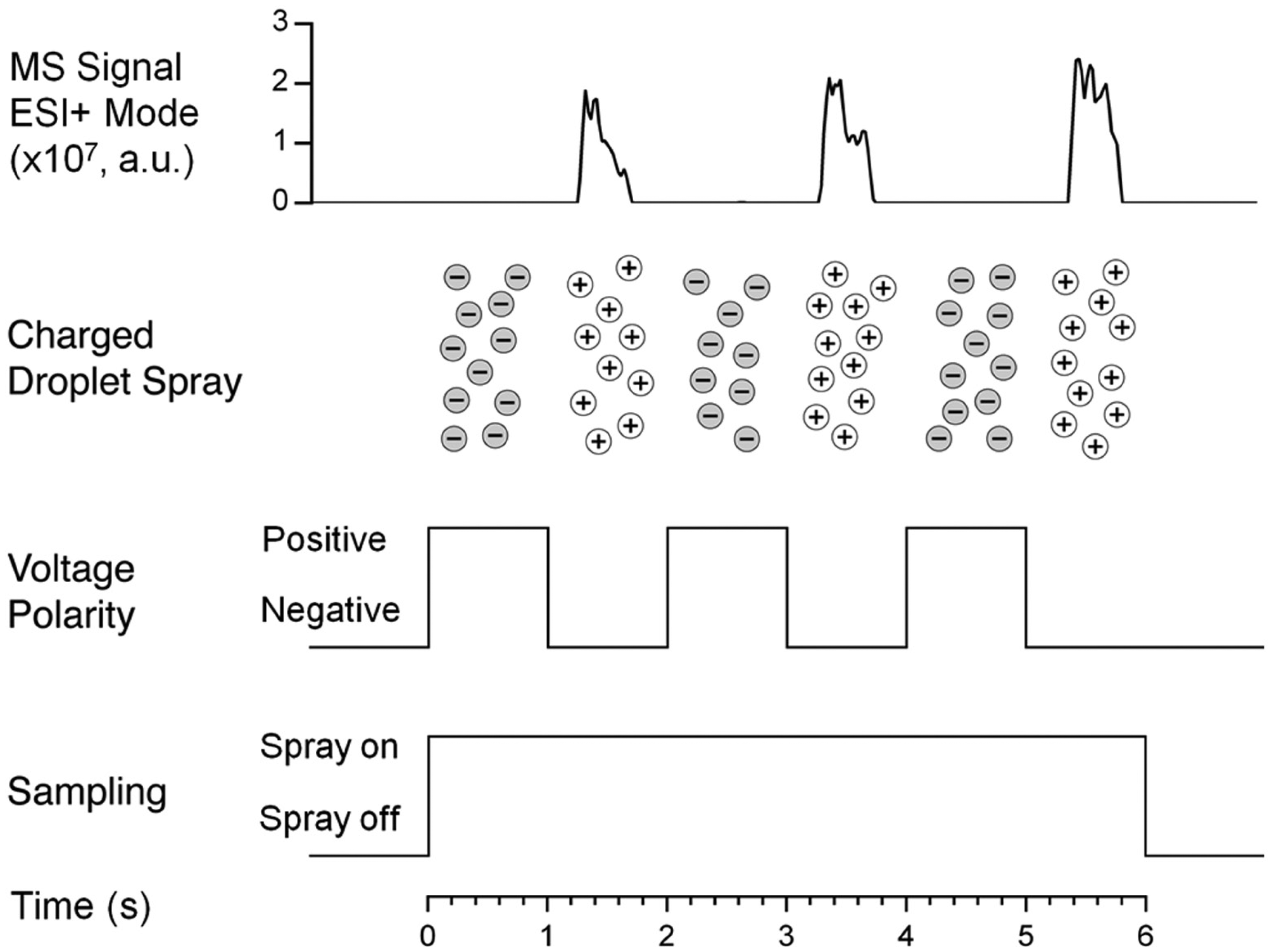
Timing schematic illustrating acoustic sampling, voltage polarity switching, ion generation, and MS signal. A 6 s burst of a 60 µM glu-fib signal generated via this process shows how high-voltage polarity is toggled between positive (generation of negative charge; no signal) and negative (generation of positive charge; observed signal). During the burst, the voltage switches polarity six times, and since the MS is set to detect ions of one charge type, we see three peaks.
To further sample the working range of this platform across chemical species types, solutions of 100 µM caffeine (data not shown), 100 µM warfarin ( Fig. 6 ), and 60 µM ubiquitin ( Fig. 7 ) were acoustically ejected into the MS, respectively. Both the caffeine and warfarin signals were approximately 20- to 30-fold lower than the MS instrumentation fitted with an optimized electrospray ionization (ESI) probe (data not shown). Warfarin generated signal in both the positive ( Fig. 6a ) and negative ( Fig. 6b ) modes. There was around a twofold loss in sensitivity between positive and negative modes for warfarin, which is similar to the loss observed using a standard ESI interface. Sampling a solution of 60 µM ubiquitin, a small 10 kDA protein produced a mass spectrum where the 8th to 12th charge states were the most prominent ( Fig. 7 ). More importantly, the distribution of this charge envelope could be driven to higher or lower charge states using the standard technique of increasing or decreasing the cone voltage (data not shown). This behavior was similar to that observed using the standard instrument ESI source.
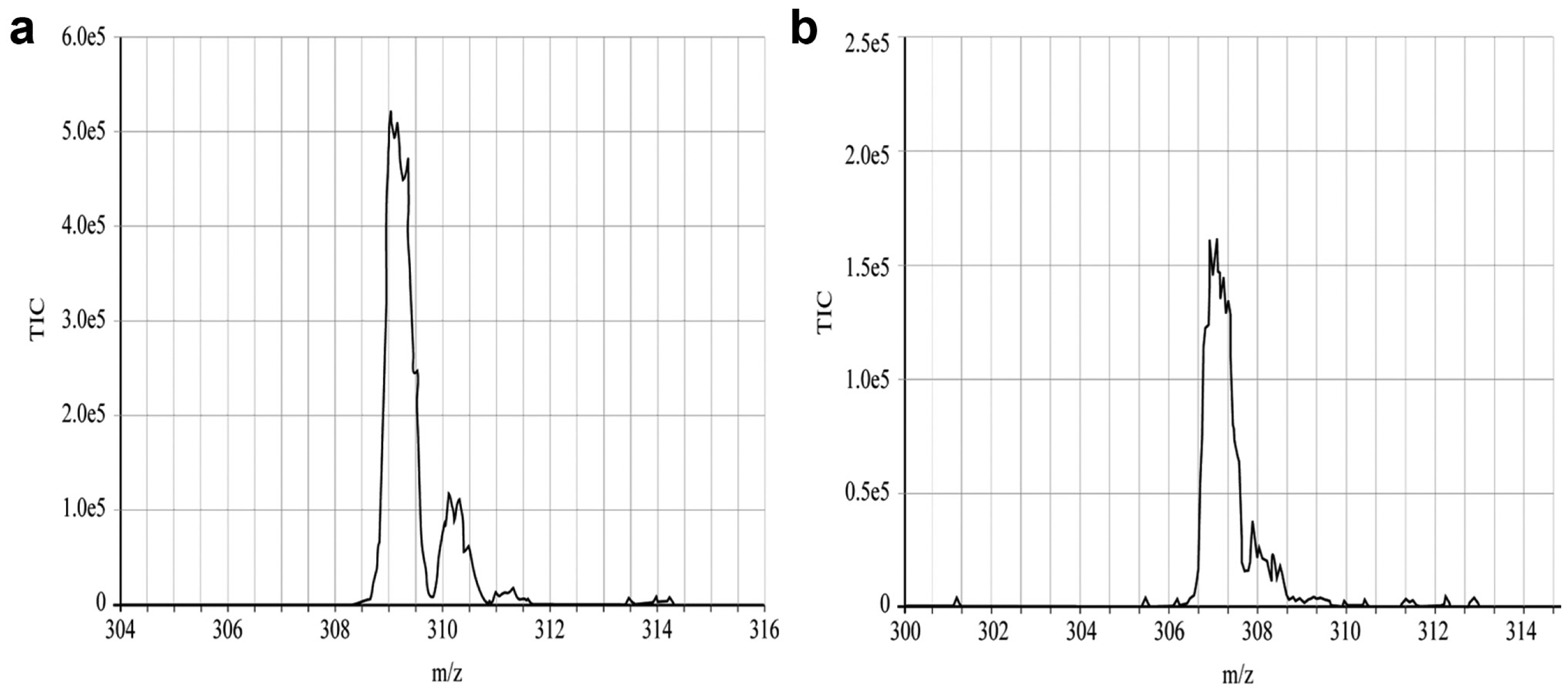
Mass spectrum of warfarin, molecular weight 308, collected on Echo-MS platform in positive (
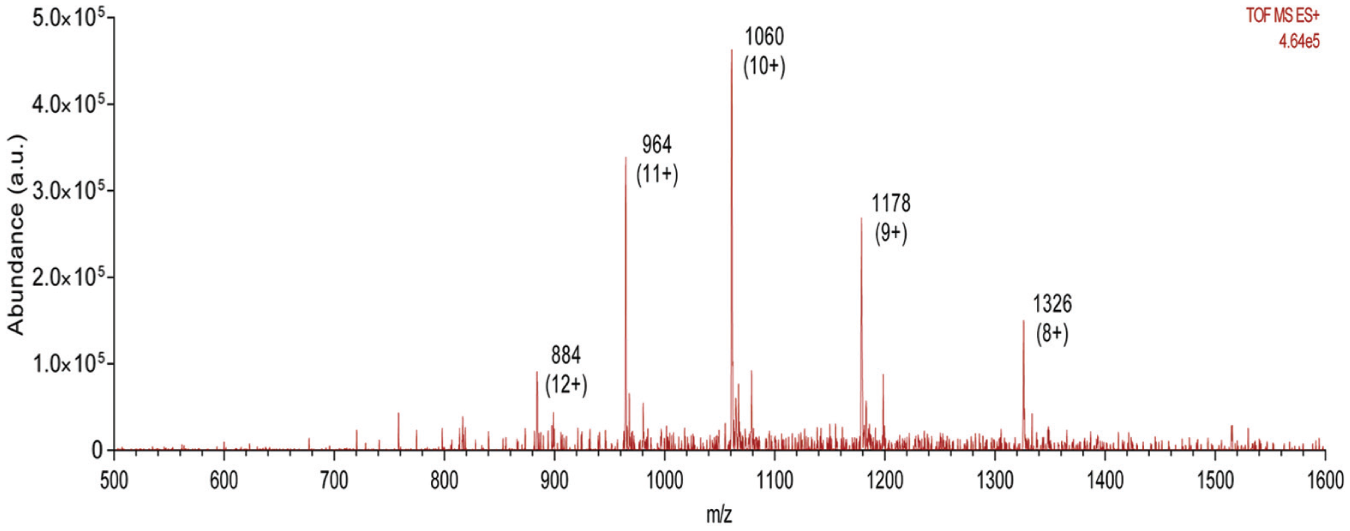
Mass spectrum of multiply charged ubiquitin protein obtained from Echo-MS platform. Annotated with the 8-, 9-, 10-, 11-, and 12-plus charge states at m/z 1326, 1178, 1060, 964, and 884, respectively. The spectrum observed is typical of that produced by commercially available electrospray sources.
The sampling speed of the Echo-MS was characterized to demonstrate its potential as a high-throughput detector for label-free assays. Signal occurs within 50 ms of the beginning of the acoustic ejection sequence and returns to baseline after the end of the ejection on a similar timescale. Signal remained relatively constant during ejection (
Fig. 8
, top), and only 1 sampling event out of the 384 did not result in a peak; this was due to a failure in the completion of the acoustic ejection in this instance. With the addition of a motorized XY stage controlled by the Echo (
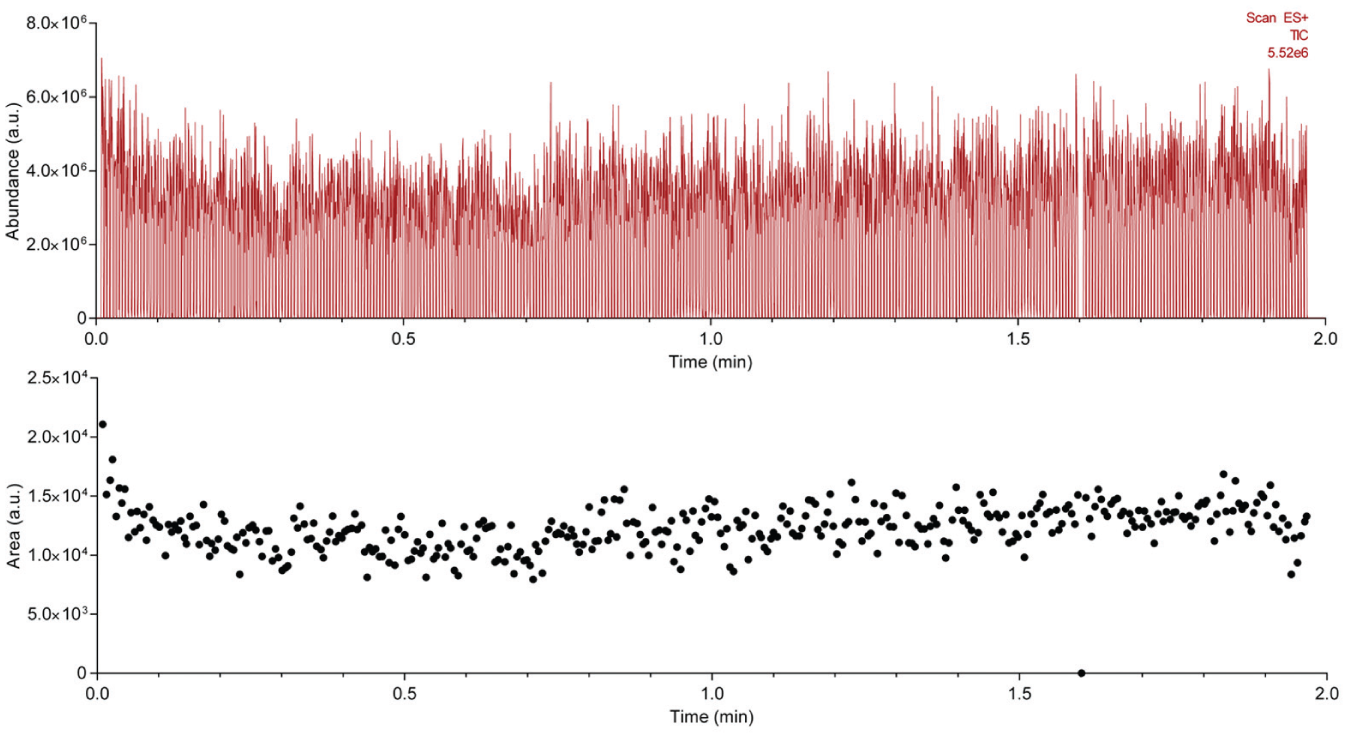
(Top) MS time trace showing 384 acoustic ejections in less than 2 min from a single well, timed to simulate the movement of the stage between neighboring wells. The data show a single failed analysis from the well at 1.6 min. These data were included as they help to add context to the speed of the analysis. (Bottom) Scatterplot of the integrated area for each of the 384 peaks, showing a CV of 14%.
The linear range of the system was investigated with a set of 12 solutions of methylated warfarin (m/z 321), ranging from 30 nM to 100 µM. In all solutions, 1 µM warfarin (m/z 307) was used as an internal standard. Triplicate rows of the solution were prepared in a 384-well PP plate, and 16 replicates of each set were acoustically ejected for 0.5 s per well. Data were collected in negative ion mode on the mass spectrometer, and a single centrioded spectrum was produced for each ejection. The log of the ratio of the peak heights of methylated warfarin (m/z 321) to warfarin (m/z 307) was plotted against the log of the concentration of methylated warfarin ( Fig. 9 ). The resulting curves demonstrate that the current prototype possesses a linear range of at least three orders of magnitude, from 0.1 µM to 100 µM, with good inter- and intrawell reproducibility.
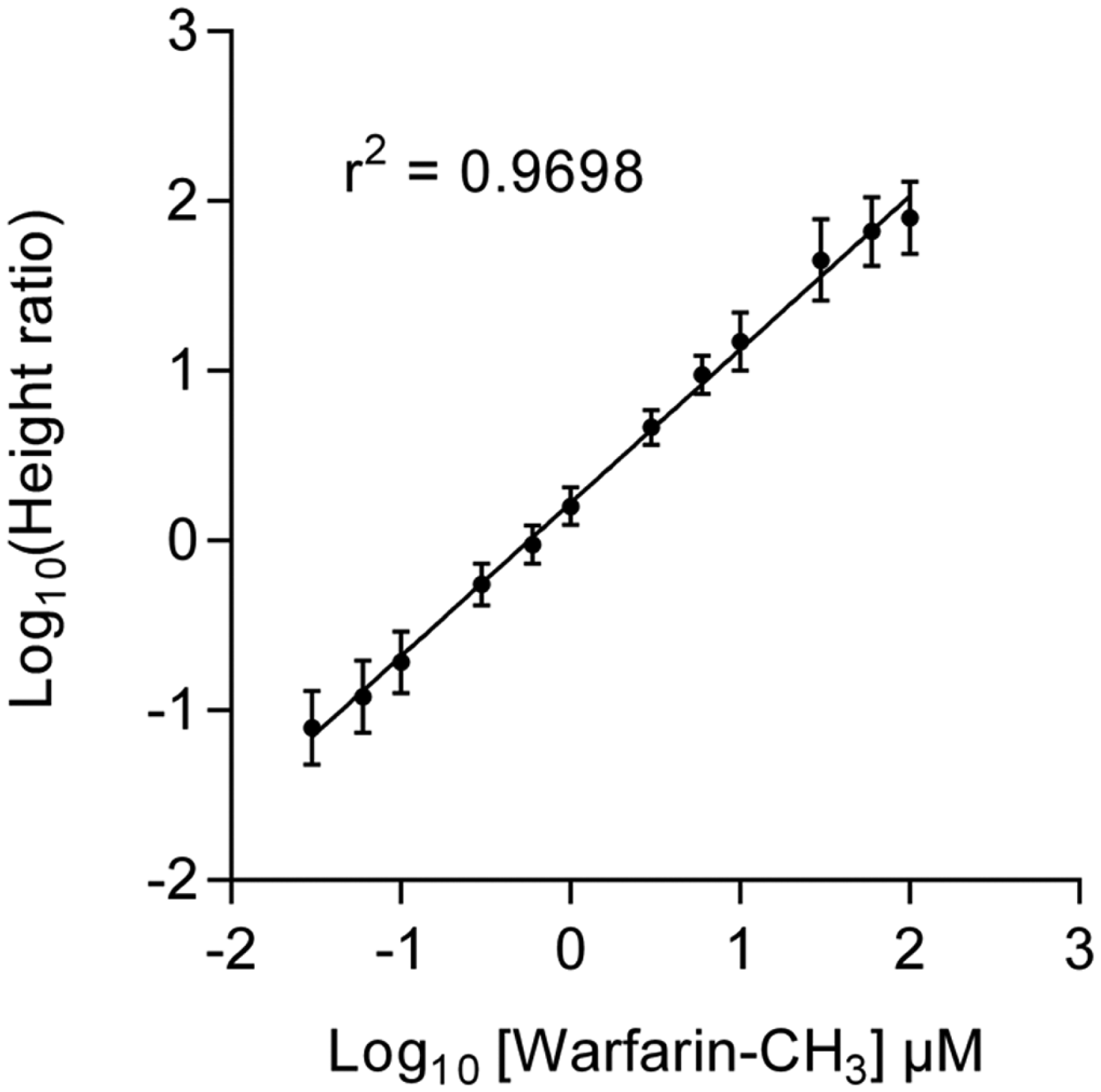
Negative ion MS signal response of warfarin-CH3, utilizing 1 µM warfarin as an internal standard, over a range of concentrations from 30 nM to 100 µM. The log of the ratio of the peak heights of methylated warfarin (m/z 321) to warfarin (m/z 307) was plotted against the log of the concentration of methylated warfarin. Each data point is generated from a 0.5 s acoustic ejection from the well, and the error bars show the standard deviation across the multiple data points. These data show good inter- and intrawell reproducibility over a linear range of three orders of magnitude.
Conclusions and Discussion
Our hybrid Echo-MS instrumentation has shown that quantitative and qualitative high-throughput mass spectroscopic analysis of samples directly from commercially available assay plates via acoustic ejection is within reach. The system is able to generate ion beams from typical test analytes with characteristic ESI spectra. Using a standard 384-well COC plate, it is possible to generate a signal from as little as 3 µL of material. With the motorized plate stage, samples can be loaded at a rate of close to three per second. This rate of sampling enables the acquisition of >10,000 data points per hour, which would be sufficient to support high-throughput screening and IC50 determination. This sampling speed may also enable mass spectrometry to be more widely used for kinetic studies.
The ESI interface developed to access the acoustically generated spray ensures the system will have wide application in cell metabolites, small-molecule drugs, peptides, and other biomolecules. However, one limitation of this system is the lack of separative technology to reduce potential matrix effects. 20 Work is currently underway to understand the potential impact of matrix suppression on biochemical screening applications and the link between droplet size and population on the ability to ionize a variety of sample molecules. In addition, future development and optimization of both the acoustic mechanisms and transfer interface can only enhance the sensitivity, reproducibility, and complexity of matrices from which samples can be detected. Ion mobility separations can be performed within the mass spectrometer to add yet another dimension to the data produced, which will become key for analysis of more complex samples.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.