
Abstract
Measuring binding affinities and association/dissociation rates of molecular interactions is important for a quantitative understanding of cellular mechanisms. Many low-throughput methods have been developed throughout the years to obtain these parameters. Acquiring data with higher accuracy and throughput is, however, necessary to characterize complex biological networks. Here, we provide an overview of a high-throughput microfluidic method based on mechanically induced trapping of molecular interactions (MITOMI). MITOMI can be used to obtain affinity constants and kinetic rates of hundreds of protein–ligand interactions in parallel. It has been used in dozens of studies to measure binding affinities of transcription factors, map protein interaction networks, identify pharmacological inhibitors, and perform high-throughput, low-cost molecular diagnostics. This article covers the technological aspects of MITOMI and its applications.
Introduction
Biological systems rely on the recognition and interaction of an immense number of biomolecules. Quantitating the interaction of individual molecules is critical to understanding cellular function, 1 developing new drugs, 2 improving enzyme activity, 2 or computationally simulating a cell, 3 to name a few. Although qualitative data have contributed significantly to our understanding of biological systems, the necessity of quantitative data has increased with the rise of systems and synthetic biology. 4 The transition from a descriptive to a predictive science is, however, hindered in part because of a lack of tools to collect quantitative data with sufficient accuracy and throughput. 4b
Molecularly, the basis for the interaction between two biological molecules is a combination of noncovalent bonds (i.e., electrostatic, van der Waals, and hydrogen) or hydrophobic interactions,2,5 yet much of the molecular behavior between two biomolecules can be accounted for by two biochemical parameters: the strength of binding (affinity) and the kinetics of reaction (association and dissociation rates). Binding force is a less frequently used parameter, given the technical difficulties to obtain it, 6 but it can provide complementary information such as the specificity of the molecular interactions, 7 and resolve low- and high-affinity interactions. 8 Nevertheless, affinity and binding rates are the principal thermodynamic properties that characterize molecular interactions. 9
Many affinity biosensors have been developed to obtain these parameters, including force-based techniques 10 (atomic force microscopy, shear flow, optical tweezers, centrifugation, and microcantilever), electrical (nanowires), 11 acoustic (quartz crystal resonators), 12 calorimetric, 12 and optical methods [surface plasmon resonance (SPR) 13 and fluorescence correlation spectroscopy]. 14 A commonly used technique to characterize kinetic interactions is SPR, 13 which is commercially available. But all of these techniques generally lack the throughput needed to deliver the data required to comprehensively characterize complex biological systems. Although efforts are moving in this direction, throughput still remains rather limited. For example, Biacore’s Flexchip can measure the interaction between one target and 400 spotted ligands in 2 h, but it requires 1.6 mL of sample. The ProteOn from BioRad generates kinetic data for up to 36 molecular interactions simultaneously. A recent SPR-based microfluidic chip showed potential to measure binding events for up to 264 different ligands against multiple analytes, yet it only measured one sample at a time per chip. 15 High-throughput sequencing–fluorescent ligand interaction profiling (HTS-FLIP) is a new technique that has been developed to study protein–DNA interactions, and it can determine disassociation constants of one transcription factor to millions of DNA targets. 16 RNA on a massive parallel array (RNA-MaP) is another recent high-throughput technique used to measure the binding affinities and disassociation rates of a single, fluorescently labeled protein to >107 RNA targets. 17 However, both HTS-FLIP and RNA-Map require a second-generation DNA-sequencing instrument. It is thus clear that technologies with much higher throughput, better sensitivity (low to high affinities and transient interactions), and more versatility (diversity of applications) are needed to study biological systems.
In this article, we discuss a high-throughput microfluidic platform capable of measuring both affinity and kinetic rates. The method is based on mechanically induced trapping of molecular interactions (MITOMI), 18 which dramatically increases throughput and sensitivity compared to other state-of-the-art techniques. MITOMI was originally developed to map the binding affinities of proteins to various DNA target sequences, but MITOMI applications have since been expanded to measure a broad range of molecular interactions, including protein–RNA, 19 protein–protein, 20 antibody–protein, 21 and protein–small molecule 22 ( Table 1 ). The technology was recently applied to obtain reaction rates of protein–protein 21 and protein–DNA interactions, 4c and it has been adapted to perform immunoassays for diagnostics. 23 Aside from being used to study protein–ligand interactions, MITOMI has also been adapted to high-throughput force-based measurements 24 and surface-patterning applications. 25
Summary of the Different Designs and Applications of MITOMI and Its Derivatives That Evolved over Time.
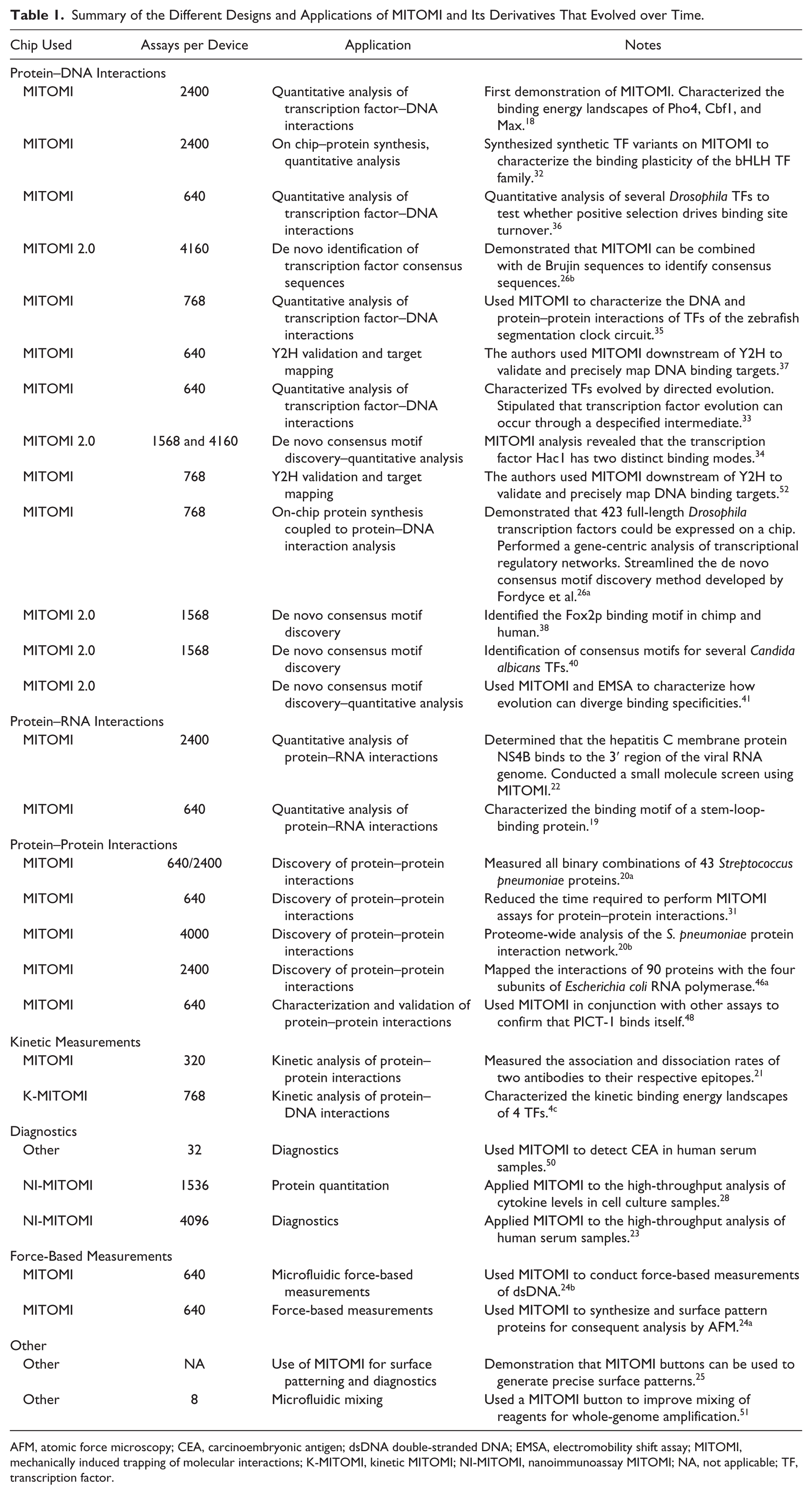
AFM, atomic force microscopy; CEA, carcinoembryonic antigen; dsDNA double-stranded DNA; EMSA, electromobility shift assay; MITOMI, mechanically induced trapping of molecular interactions; K-MITOMI, kinetic MITOMI; NI-MITOMI, nanoimmunoassay MITOMI; NA, not applicable; TF, transcription factor.
MITOMI
The MITOMI platform combines two different techniques, microarrays and a new microfluidic detection mechanism (the actual MITOMI process). A MITOMI microarray consists of a planar substrate, usually an epoxy-coated glass slide, onto which minute amounts of biological solutions (~1–10 nL) are printed using standard DNA microarray robots. Slides can contain thousands of spots of biological solutions, including DNA,4c,18,20a,26 RNA,19,22 small molecules,22,27 cell culture media, 28 human serum, 23 antibodies, and live cells. 29 Once spotted, a microfluidic chip is aligned and bonded to the microarray ( Fig. 1a ).
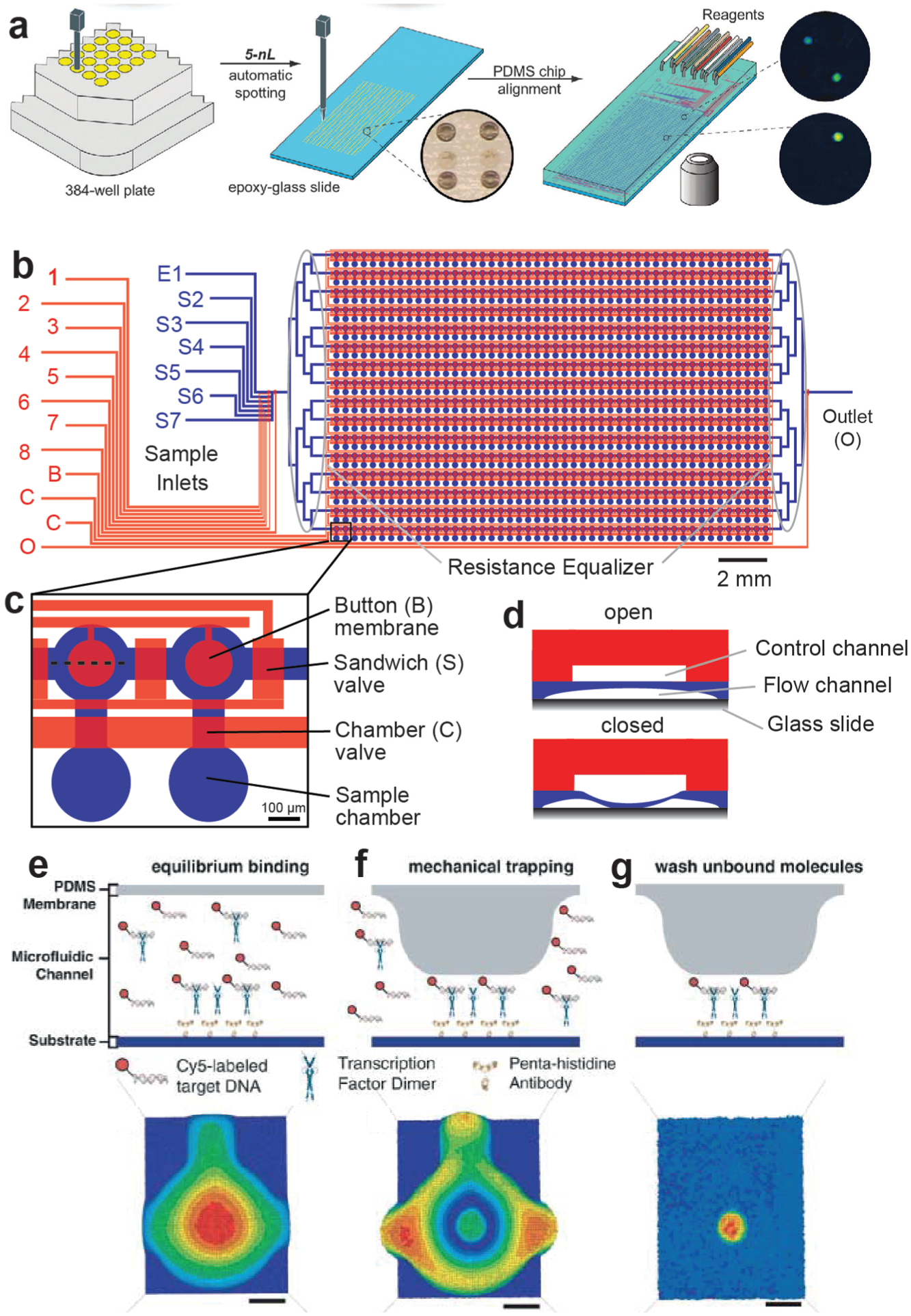
Mechanically induced trapping of molecular interactions (MITOMI) workflow. (
Microfluidic devices are fabricated by multilayer soft lithography 30 using poly-dymethilsiloxane (PDMS). MITOMI chips generally consist of two layers: a flow layer, which is in contact with the substrate and contains a network of microfluidic channels to perform the biological assays, and a control layer, which comprises MITOMI button membranes and valves to control fluid flow ( Fig. 1b ).
The core of a MITOMI chip is a microfluidic unit cell composed of a spotting chamber and a detection area separated by a “neck” valve. The number of microfluidic unit cells in a MITOMI chip can range from 640 to 4160 units. The spotting chamber compartmentalizes the printed biological solution. “Sandwich” valves isolate unit cells from each other during incubation steps to eliminate cross-contamination. Inside the detection area lies one or more button membranes that, when actuated, contact the glass surface of the microchannel, covering a circular area but not completely blocking flow through the channel (
Fig. 1C
An experiment begins by preparing the surface chemistry using standard biotin–avidin chemistry. Generally, biotinylated bovine serum albumin (biotin-BSA) is flowed through the chip with the spotting chambers closed, coating the detection area, followed by Neutravidin. Next, the button is actuated, and biotin-BSA flushed again through the chip. At this point, the surface beneath the button is covered by Neutravidin, whereas the rest of the channel is passivated with biotin-BSA. With the button released, biotinylated proteins or antibodies can be introduced and selectively bound to the area below the button membrane. Subsequent biochemical steps depend on the given application and are discussed in more detail in the next sections of this article. One potential caveat of MITOMI is that it is a fluorescent-based technique: the molecule of interest must be conjugated to one or more fluorescent dye molecules to detect the molecular interaction. An automated microarray fluorescent scanner images the whole chip to measure the signals from the ligand–protein complexes formed beneath each button membrane.
Measuring Molecular Affinities with MITOMI
Transcription Factor–DNA Interactions
Many molecular interactions exhibit fast off-rates and medium to low affinities (nM to µM), but high-throughput techniques often miss these transient and low-affinity interactions. MITOMI was originally developed to fill this gap. The first application of the technology was the measurement of binding affinities of transcription factors (TFs) to hundreds of different DNA sequences. 18 The operation for the chip is as follows ( Fig. 2 ): first, Cy5-labeled double-stranded DNA target sequences are printed as a microarray, which is then aligned to the MITOMI device. Each sequence is spotted at several concentrations to later obtain a saturation binding curve and derive absolute binding affinities. Next, anti-penta histidine antibodies are immobilized under the button while a solution of in vitro transcription–translation (ITT) reaction mix, which includes a DNA template coding for the his-tagged TF protein, is prepared off-chip and boron-dipyrromethene (BODIPY)-labeled tRNA is added to the mixture for protein labeling. The BODIPY-labeled proteins are loaded into the chip, where they bind to the anti-his antibodies immobilized beneath the buttons. Then, the button is pressurized to protect the antibody–protein complex while the neck valve is opened to permit the solution to enter the spotting chamber, where it solubilizes the spotted DNA. Once all spotting chambers are filled with liquid, the neck valve is closed again, and the detection chambers are washed to remove any solution phase molecules. The sandwich valves are closed, and the solubilized DNA is allowed to interact with the TFs. When equilibrium is reached, after ~1 h incubation, the chip is scanned to quantitate how much Cy-5-labeled DNA is in solution within each chamber. Next, antibody–TF–DNA complexes are protected with the button membrane. The neck valve is once again closed, the sandwich valves are opened, and any unbound material in the detection area is washed away. This is followed by a second and final fluorescent scan that quantitates how much BODIPY-labeled TF is present in each MITOMI detection area and how much CY-5-labeled DNA target is bound by these TFs. This whole process requires ~4–5 h, but by changing the architecture and chemistry of the original MITOMI design, it is possible to reduce assay times down to 2.5 h. 31
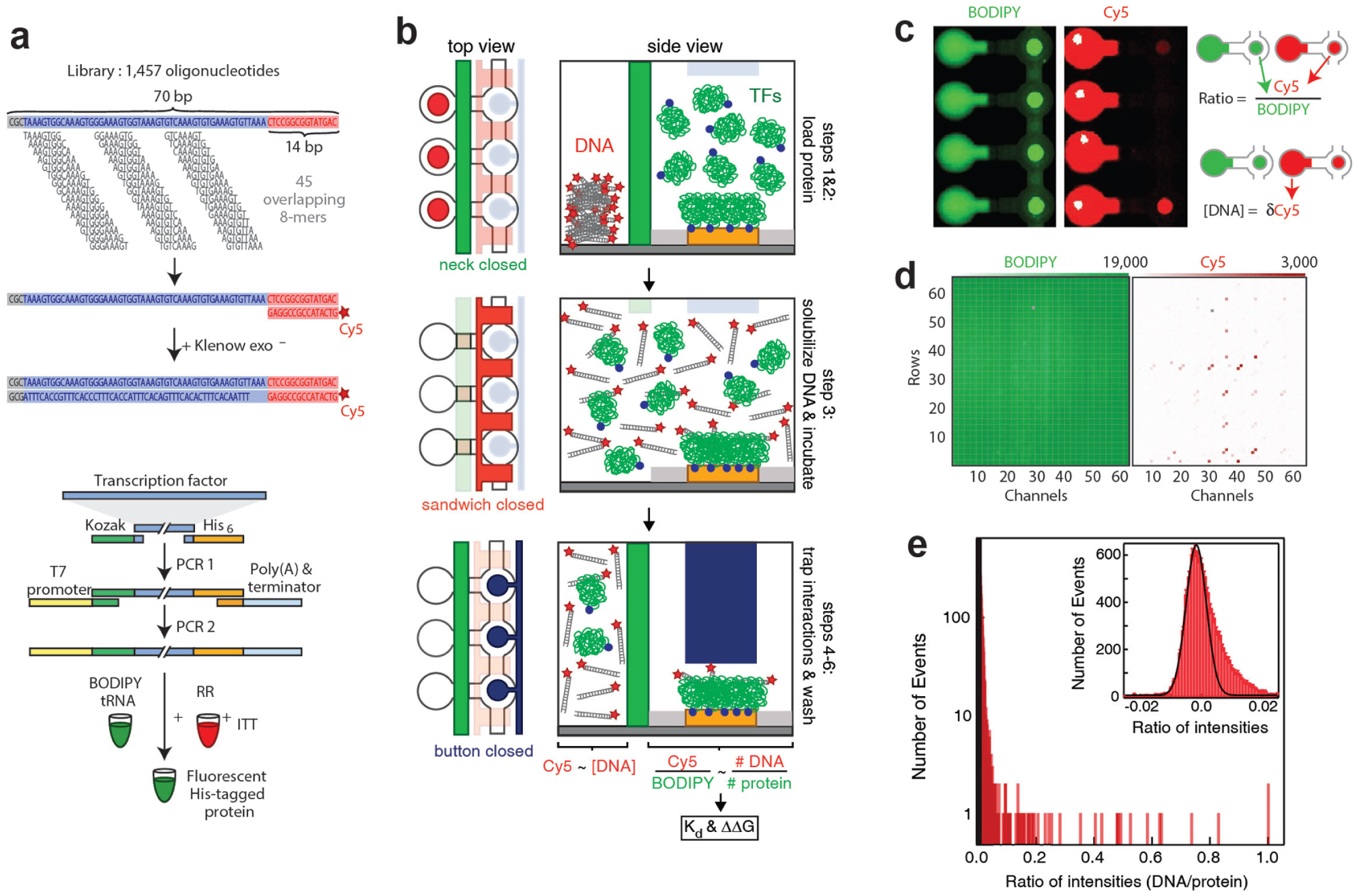
High-throughput measurements of binding affinities of protein–DNA interactions using mechanically induced trapping of molecular interactions (MITOMI). (
Affinities are determined from the amount of surface-immobilized TF, the amount of DNA bound to the protein, and the amount of solution phase DNA. Therefore, only two sites of each microfluidic unit cell are analyzed. The first site is the area under the button membrane, which contains the molecular complex where the BODIPY fluorescent intensity represents the amount of surface-bound TF protein molecules and the Cy5 intensity reflects the amount of DNA molecules bound by those TFs. The second site is the spotting chamber, where CY5 intensities represent the concentration of unbound DNA. With a DNA calibration curve, it is possible to calculate the equilibrium dissociation constant of the TF to the DNA, Kd, using a single-site binding model with the expression r = (rmax × [D]) / ([D] + Kd), where r represents the fractional occupancy (bound DNA/protein); [D] the free DNA concentration; and rmax the value at which all TF binding sites are occupied, leading to saturation.
Using MITOMI, Maerkl and Quake 18 studied the interactions of four TFs belonging to the basic helix–loop–helix family (bHLH) to 464 DNA targets. Each MITOMI chip measured 2400 interactions in parallel. Using seven microfluidic chips, the authors obtained more than 41,000 data points, which allowed them to map the binding energy landscape for these eukaryotic TFs. This study provided the first comprehensive and quantitative binding energy landscape of any transcription factor, and it improved existing consensus sequences for these transcription factors. Following this work, the same authors studied the binding plasticity of the basic bHLH family by measuring the binding specificities of 95 MAX TF mutants. 32 In an another study, Shultzaberger et al. used MITOMI in combination with quantitative PCR and binding site fitness assays to characterize the binding, function, and fitness of evolved variants of the helix-turn-helix transcription factor MarA, and they stipulated that TFs could evolve by traversing through a despecified intermediate. 33 Fordyce et al. 26b measured the binding affinities of 28 Saccharomyces cerevisiae transcription factors to a library of 1457 double-stranded oligonucleotides to obtain de novo consensus sequences. The authors were able to measure DNA-binding specificities for TFs that were difficult to obtain with other state-of-the-art techniques. The same group studied the interactions of different genomic target sites to the transcription factor Hac1, a basic leucine zipper (bZIP) TF involved in the unfolded protein response. 34 The authors also showed that Hac1 binds at least two different target sites of different lengths.
MITOMI was instrumental in the characterization and description of a new circuit model of the zebrafish segmentation clock. 35 Characterization of DNA binding activity and protein–protein interactions of transcription factors were performed with MITOMI. Using MITOMI, He et al. carried out a quantitative analysis of several Drosophila TFs to test whether positive selection drives binding site turnover. 36 To automate the screening of proteins that bind to Drosophila melanogaster regulatory elements, Hens et al. developed a high-throughput yeast one-hybrid platform, which involved the usage of MITOMI to validate previously unidentified TF–DNA interactions. 37
More recently, Nelson et al. found that human FOXP2, a transcription factor believed to be important in language evolution, has almost identical binding profiles to the chimp FOXP2. 38 MITOMI also played a role in the identification of a new transcription factor (White-Opaque Regulator 3) 39 and in the description of a circuit responsible for the white-opaque switch 40 in Candida albicans, the most common fungal pathogen of humans. The same group also showed that successive duplications of transcription factors acquired a distinct group of target genes by gaining different DNA-binding specificities, different preferences for half-site arrangements, and different associations with cofactors. 41
Rockel et al. developed an approach to characterize gene regulatory networks based on MITOMI 26a called integrated systems-level interaction mapping (iSLIM) of TF–DNA interactions. iSLIM synthesizes hundreds of full-length transcription factors on chip, followed by biophysical characterization with target DNA. The authors characterized the interaction of 423 full-length Drosophila TFs with 12 DNA sequences and determined kinetic rates, affinities, specificities, and consensus sequences.
Protein–Protein Interactions
Protein interactions mediate a plethora of biological processes. Some of these interactions, such as antibody–antigen, protease–inhibitor, and receptor–ligand interactions, are of significant interest to the pharmaceutical industry. Aberrant protein–protein interactions have been associated with various diseases such as Alzheimer’s, some forms of rheumatoid arthritis, spongiform encephalopahties, and familial amyloidotic polyneuropathy. 42 In addition, understanding and characterizing how proteins interact in an organism are major tasks in systems biology. 20a
So far, protein–protein interactions have been qualitatively characterized with biochemical, biophysical, and genetic techniques, including the yeast two-hybrid method, 43 mass spectrometry, immunoprecipitation, correlated mRNA expression, and protein binding microarrays.26a,44 However, the speed at which proteins are discovered or predicted demands accessible and affordable high-throughput techniques. 45 MITOMI has emerged as a viable alternative, as demonstrated by recent studies.20,26a,46
To study protein–protein interactions with MITOMI, the operation of the chip is slightly modified as compared to in protein–DNA studies ( Fig. 3 ). Instead of one oligo, DNA templates (coding for both bait and prey proteins) are spotted on the slide. A capture antibody that recognizes the bait protein is immobilized under the button membrane, followed by on-chip protein expression using an in vitro transcription–translation reaction mix. Fluorescently labeled antibodies against prey and bait proteins are flowed in the chip and incubated with the proteins, where interactions are determined from fluorescent intensities. This mode of MITOMI was coined protein interaction network generator (PING), 20a and (1) it allows for the expression of thousands of protein combinations on a single device; (2) there is no need for protein purification; (3) each reaction is isolated, preventing cross-contamination; (4) and only 2 nL of reagent is consumed per reaction, which significantly reduces cost. Proteins within a range of 37–757 amino acids are expressed on chip, although expression levels can differ up to fourfold.
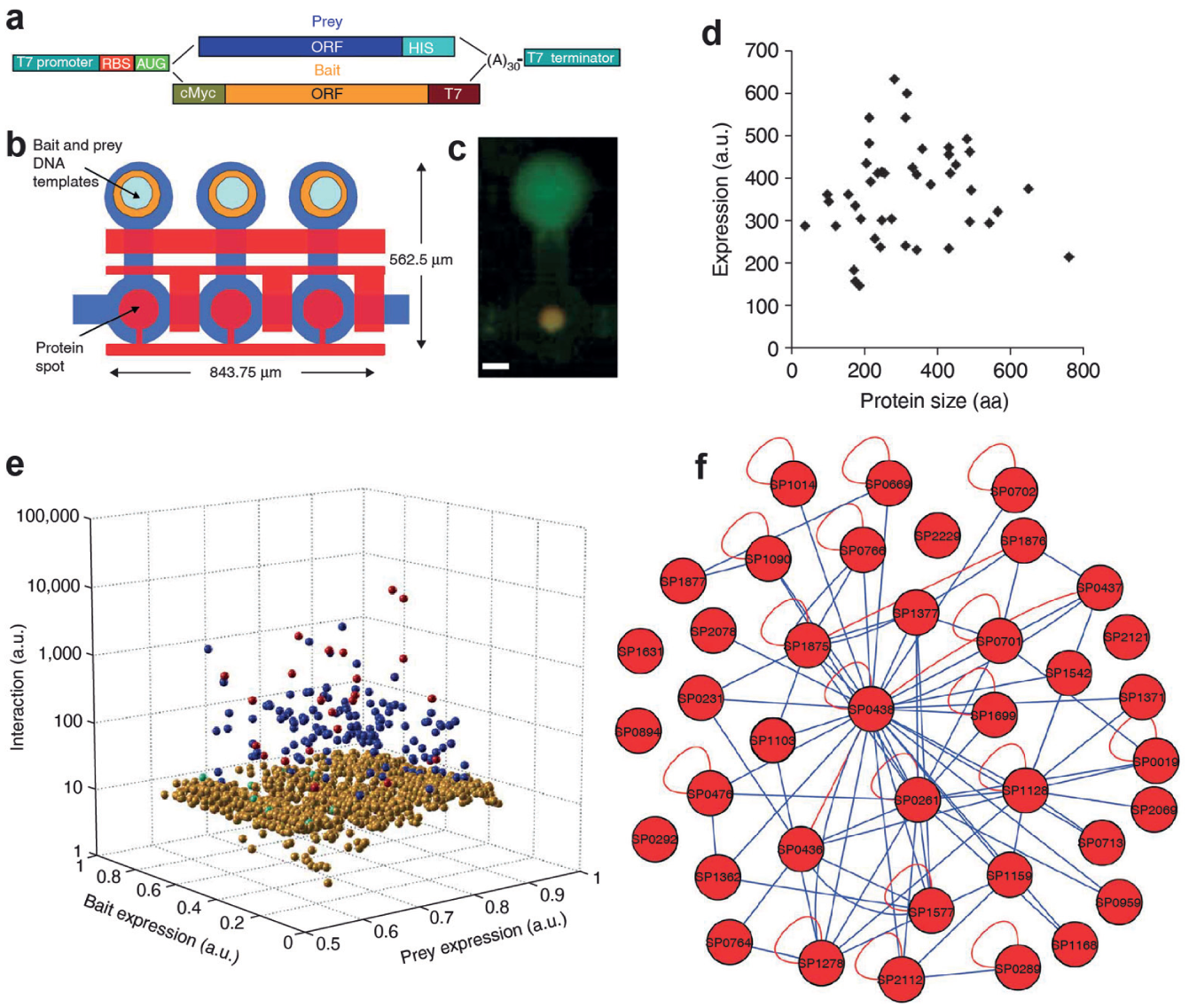
Usage of mechanically induced trapping of molecular interactions (MITOMI) as a protein–protein interaction tool. (
Using PING, Gerber et al. characterized the protein interaction network of 43 Streptococcus pneumoniae (SP) proteins. 20a They measured protein expression and all possible pairwise interactions among these proteins, finding 32 homodimers, six heterodimers, and five monomers. The resulting network consisted of 43 nodes and 157 edges. Notably, new protein interactions were discovered by applying PING. Meier and colleagues elaborated on this work and created a physical interaction map of proteins of unknown function of the S. pneumonia strain TIGR4. 20b The study led to the identification of 163 new protein interactions, which had not been reported in other data repositories, and assigned putative functions to 50 conserved proteins of unknown function. PING has also been used to study viral–host interactions43,47 and the subunits of E. coli RNA polymerase. 46a More recently, PING, in conjunction with other techniques, helped reveal direct self-association and formation of homo-oligomers of the human protein interacting with carboxyl terminus 1 (PICT-1) protein, which has been suggested to function either as an oncogene or as a tumor suppressant. 48
Running MITOMI chips can require up to 7 h, which includes 4.5 h for surface passivation and antibody immobilization, 1.5 h for protein expression, 0.5 h for protein pull-down, and 0.5 h for detection. In an effort to shorten this time, Meier et al. reengineered the architecture of MITOMI and established a new surface chemistry that decreases the operation time to 2.5 h. 31 In the new chip design, protein expression and surface passivation occur in parallel, resulting in a reduction of 4.5 h.
Binding Kinetics
Another parameter of interest describing a molecular interaction is the kinetic rate. MITOMI was adapted to determine the association and disassociation rates of an antibody–antigen interaction. 21 However, this initial demonstration remained limited in throughput. Geertz et al. further improved the performance of the microfluidic chip to measure up to 768 independent kinetic rates in parallel 4c ( Fig. 4 ). New features were added to the original MITOMI design to maximize button actuation speed, including a fluidic capacitor in the spotting chamber for pressure relief during button closure and a reduction of the fluidic resistance leading to the button membranes. This new device, also called kinetic MITOMI, or k-MITOMI, measures off- and on-rates by opening the button membrane for brief periods of time.
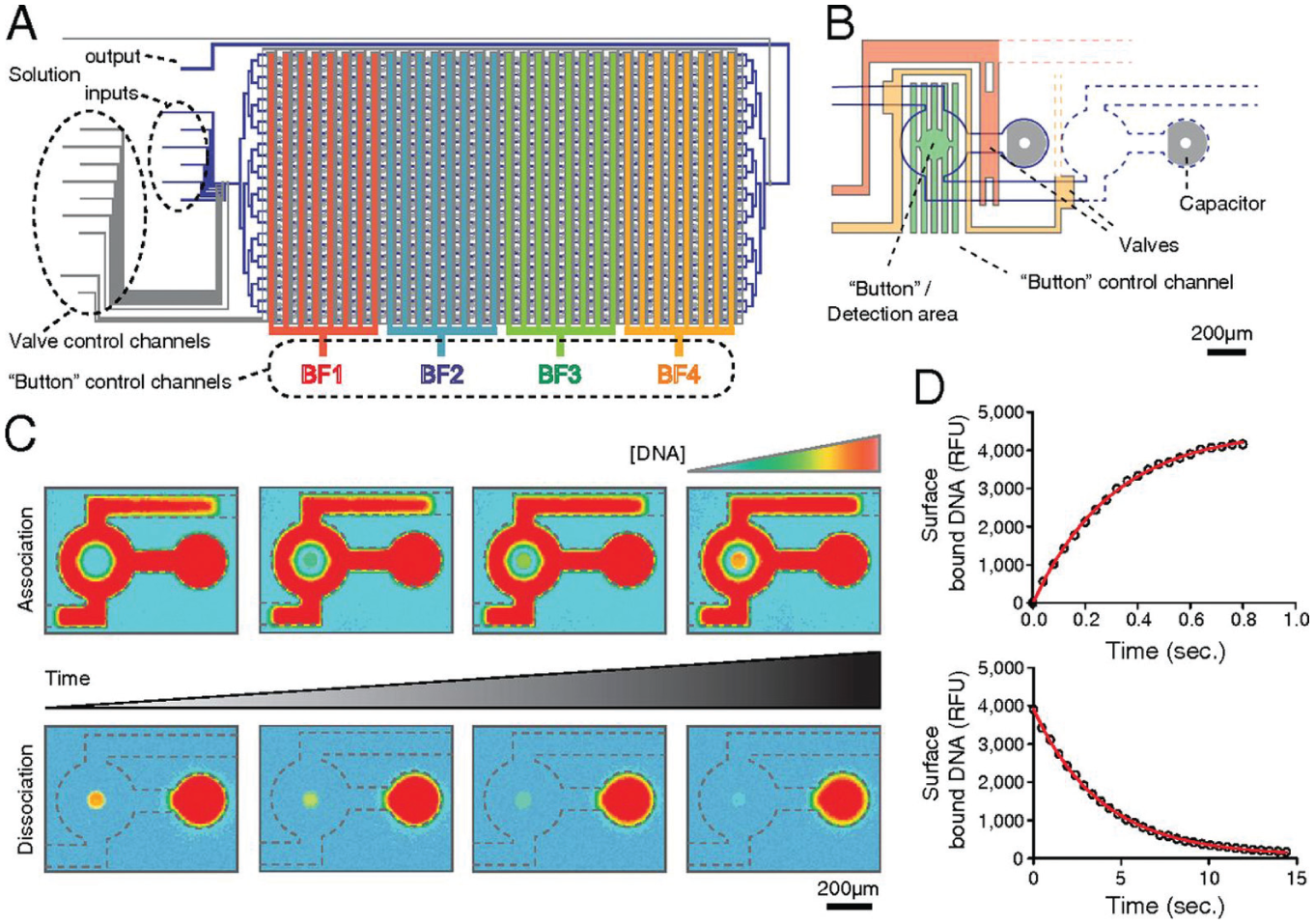
The Kinetic MITOMI (mechanically induced trapping of molecular interactions) Platform.
4c
(
To obtain an association curve, pulses start with the button closed. Solvated fluorescently labeled ligands are allowed to diffuse from the spotting chamber into the assay unit and allowed to equilibrate. Next, the button membrane is opened, and ligands bind to the surface-bound proteins. After a period of time, the button is closed, trapping protein–ligand complexes formed on the substrate beneath the button. Every time the button is open, more ligands bind to the protein until equilibrium is reached. The entire chip is imaged to determine the amount of ligand bound to the proteins, with fluorescent signal intensity being proportional to the amount of DNA bound. This process is repeated several times to construct an association curve. To obtain a dissociation curve, the surface-bound protein is initially saturated with ligands. The button membrane is pulsed (open) for a short time interval in which ligands dissociate from the surface-bound proteins. The chip is imaged every time the button is pulsed, similar to the procedure for obtaining an association curve. The minimum pulse duration of k-MITOMI is 5 ms, which allows, in principle, to measure disassociation rates as fast as ~10 s−1. 24b
To demonstrate the utility of this platform, binding kinetics for the mouse C2H2 zinc finger TF Zif268 and the S. cerevisiae transcription factors Tye7p, Yox1p, and Tbf1p to target DNA sequences were characterized. 4c In total, 684 association and 1704 disassociation curves were collected for 223 molecular interactions. This study showed that transcription factor specificity is solely determined by the dissociation rate and that association rates are quite uniform across TFs belonging to different transcription factor families.
Molecular Force Assays
Molecular force measurements provide complementary information to chemical studies of biomolecular interactions by revealing structural differences and functional aspects of the molecular complex, 49 the spatial distribution of the receptors and the antigens for immunoassays, 5a the specificity of the interactions, 7 and differences in binding mode. 7 Single-molecule force spectroscopy is a technique used to measure the binding strength between two molecules, which are usually in the order of piconewton forces. 49a Although several techniques exist, such as atomic force microscopy, optical tweezers, and magnetic tweezers, they are time-consuming, extremely low-throughput, costly, and difficult to set up.6,7,24a To address this problem, Otten et al. adapted MITOMI to measure molecular forces 24b ( Fig. 5 ). In this new configuration, neutravidin molecules are immobilized on the glass surface while two DNA duplexes are immobilized on the opposite side of the channel (PDMS surface) exactly below the button membrane. The DNA duplexes consist of a probe strand (covalently attached to the PDMS surface) and a biotinylated reference strand, both labeled with different fluorophores. The button membrane is actuated to contact the surface. On pressure release and button retraction, some biotinylated reference DNA strands would bind to neutravidin, while some of the bonds of either the reference or probe strands would cede and rupture. This bond breakage is proportional to their relative strengths. The fluorophore distribution is scanned at the top and bottom of the channel with a confocal microscope to quantify the amount of bond rupturing. As a proof-of-concept of this approach, they tested binding of EcoRI, an endonuclease enzyme, to two different binding sequences, finding that their results were consistent with previous reports.
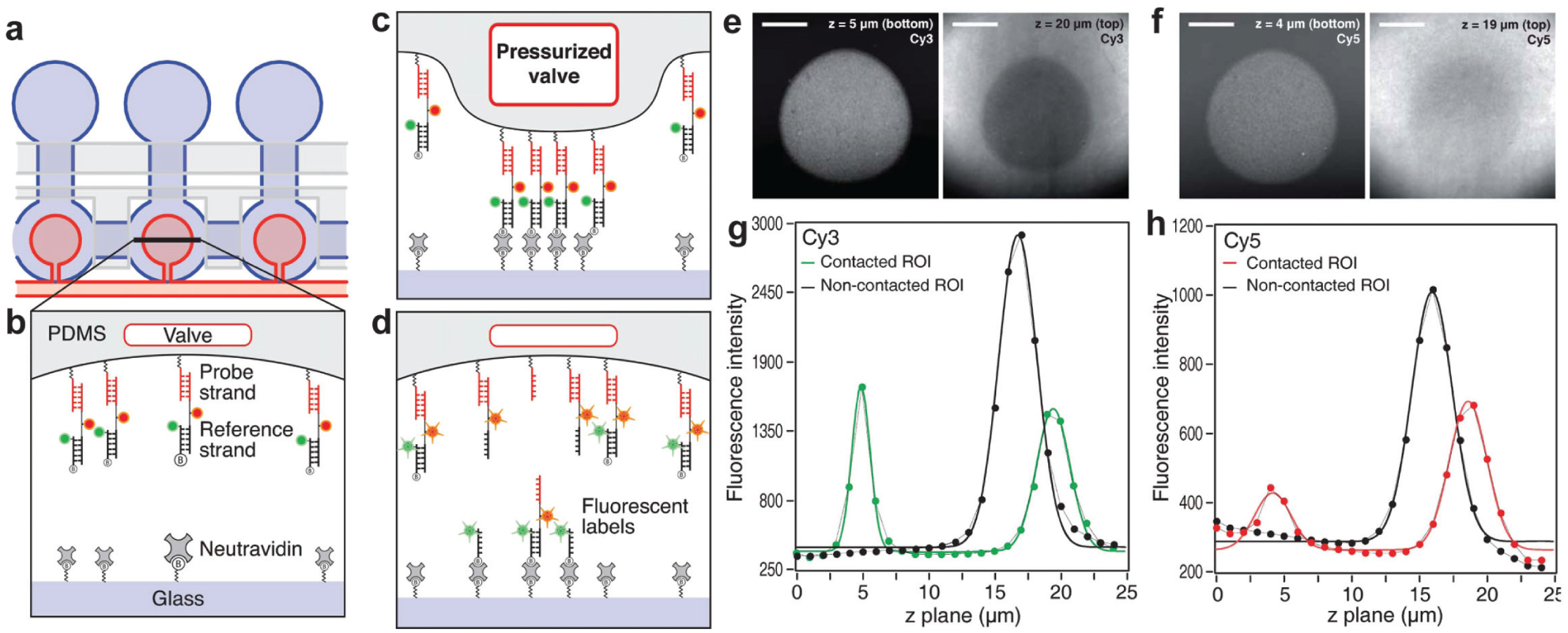
Usage of mechanically induced trapping of molecular interactions (MITOMI) as a molecular force assay. (
Another application of MITOMI was recently reported by the same group, 24a in which MITOMI was used to create a protein microarray from a spotted gene array. The on-chip synthetized proteins are covalently attached to the glass slide under each button membrane. The microfluidic device is peeled from the glass slide, the exposed protein microarray is scanned with an atomic force microscope, and single-molecule unfolding traces from each protein construct are recorded.
Immunoassays
Most MITOMI applications have been developed to study fundamental aspects of molecular interactions. Recently, we adapted MITOMI to use as a low-cost, high-throughput immunoassay platform to measure biomarker levels from different biological samples23,28,50 ( Fig. 6 ). The modified MITOMI device comprises bigger spotting chambers (5 nL) than the original MITOMI devices. This increase in volume allows for the simultaneous detection of four different biomarkers per sample. We showed that sensitivity is comparable to enzyme-linked immunosorbent assays (ELISA), while reducing volume and reagent cost by at least 1000-fold.23,28 The first generation of these devices could analyze up to 384 different samples for a total of 1536 assays. The device was used to assess the synergistic action of binary adjuvant cocktails by measuring the production of four inflammatory cytokines (IL-6, TNF-α, IL-12p70, and IL-23) from dendritic cells. 28 Cells were stimulated with 435 different binary combinations of 10 adjuvants that targeted members of the toll-like receptor family. The study revealed several synergistic adjuvant combinations, which might lead to the development of more effective vaccines. These combinations were further evaluated in a mouse model.
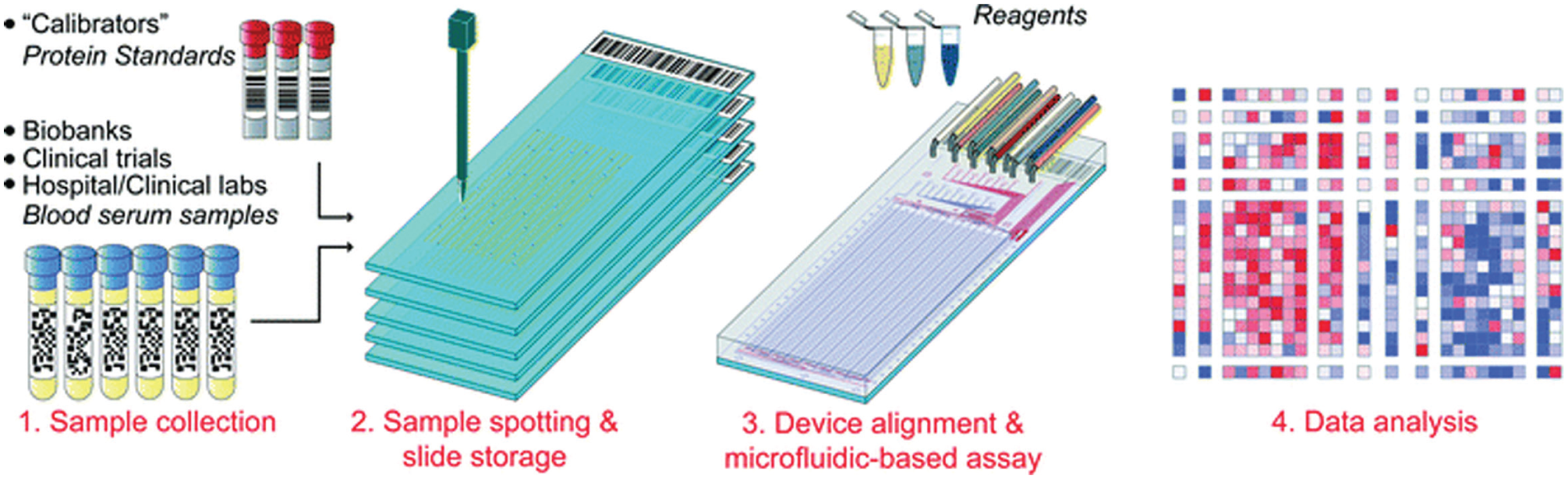
Usage of mechanically induced trapping of molecular interactions (MITOMI) to measure biomarkers in serum samples. Up to 1024 different serum samples collected from biobanks, clinical trials, or clinical laboratories can be spotted on multiple glass slides. A microfluidic device is aligned and bonded on top of the slide. The device can detect and measure up to four biomarkers for each sample for a total of 4096 assays on a chip. Reprinted from Garcia-Cordero, 23 by permission of the Royal Society of Chemistry.
A second-generation device was developed to measure up to 1024 samples in one chip for a total of 4096 assays. 23 The microfluidic device was used to measure four different biomarkers (TNF-α, IL-6, and IL-1β), including one commonly used biomarker to diagnose prostate cancer [prostate-specific antigen (PSA)] in human serum samples. The MITOMI data correlated well with ELISA-based measurements (R2 = 0.78); however, the microfluidic platform used 10,000 times less sample volume and reduced the cost of reagents by four orders of magnitude as compared to ELISA. Prior to this work, Zheng and colleagues showed that samples could be flowed through the channels instead of spotting them, and this led to similar sensitivities but reduced throughput. 50
Other MITOMI Assays
In search of new drugs capable of inhibiting the replication of the hepatitis C virus (HCV), a leading cause of hepatitis, Einav and colleagues 22 studied the binding of RNA to the transmembrane protein NS4B of the virus. In this study, MITOMI was used to (1) obtain the affinity constant for the NS4B–RNA protein interaction, (2) determine the structural motifs responsible for that interaction, and (3) carry out a small-molecule screen of 1280 compounds that could inhibit this interaction. An alternative approach to spotting RNA was demonstrated by Martin et al. 19 The authors demonstrated on-chip transcription of RNA from spotted DNA oligos and characterized the affinity of a library of transcribed single and double RNA mutants to a stem-loop-binding protein. Measurements agreed with electrophoretic mobility shift assays.
Ronen et al. studied DNA methylation in vitro using MITOMI. 27 The assay relies on immobilizing hairpin-shaped Cy5-labeled DNA probes of different methylation states under the button membrane and exposing them to different concentrations of a HPAII endonuclease. Using MITOMI, the authors also tested the activity of bacteria HPAII DNMTase, a DNA methyltransferase enzyme, to catalyze the transfer of a methyl group. Finally, to evaluate the performance of the device to small chemical compounds, they used MITOMI to assess the effect of four molecules in the inhibition of HPAII DNMTase.
Protein patterning has found many applications in biology, tissue engineering, and biosensors. 25 Generally, proteins are first immobilized on a substrate using a variety of techniques. However, once patterned, a separate microfluidic device is aligned and bonded to the substrate to perform an assay. To circumvent this problem, we reported a microfluidic technique to pattern multiple proteins within the same microfluidic device. 25 Starting with the button membrane actuated at high pressure, the first protein is introduced into the chip, followed by a washing step. The pressure is slightly decreased, and another protein flowed and washed. These steps are repeated for the rest of the proteins, giving rise to a pattern in the form of concentric annuli.
Finally, MITOMI can be used as a mixing element in microfluidic devices. Yang et al. demonstrated on-chip DNA amplification of lysed cells. 51 MITOMI was used to mix reagents from the moment a cell suspension is introduced into the chip to the point when all different reagents are sequentially loaded for single-cell, whole-genome amplification.
Conclusions
Although different techniques have been developed throughout the years to interrogate molecular interactions, throughput remains a major limitation and an open challenge. As we described in this review, MITOMI offers a solution to this problem by combining two techniques: microfluidics and microarrays. Microarray robots facilitate the transport, manipulation, and deposition of hundreds to thousands of low-volume samples (nL) to a planar substrate while microfluidics allow the encapsulation, control, and automation of each reaction. One important element, which makes the microfluidics unique, is the button membrane, which allows “freezing” of molecular interactions by trapping them between the PDMS and the glass surface, thus giving enough time to measure thousands of assays in parallel, but also facilitating washing steps and precluding contamination between assays. These characteristics and the notion that almost any protein-ligand interaction can be characterized with MITOMI has made it unique among the plethora of other microfluidic devices that measure binding affinities and kinetic rates. With a few modifications, MITOMI has also found many other interesting applications such as in force spectroscopy and cancer diagnostics.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been partially supported by CONACYT, Mexico, under Grant No. 226061.