
Abstract
Precise determination of disulfide linkages between cysteine (Cys) residues in proteins is essential in the determination of protein structure. Therefore, a reliable automated method for the identification of disulfide bridges can serve as an important tool in the analysis of the tertiary structure of proteins of interest. Here, we describe the current and past methods used to identify disulfide bridges in proteins, with a focus on mass spectrometry (MS)–based methods and a particular emphasis on nanoliquid chromatography–tandem mass spectrometry (nanoLC-MS/MS)–based methods. We also show the development of an easy method based on the separation of disulfide-linked proteins by sodium dodecyl sulfate–polyacrylamide gel electrophoresis under denaturing and nonreducing conditions and selective in-gel digestion of proteins using reducing and nonreducing conditions, followed by analysis of the resulting peptide mixture by nanoACQUITY UPLC coupled to a quadrupole time-of-flight (QTOF) Micro mass spectrometer (nanoLC-MS/MS). Data-dependent analysis (DDA) nanoLC-MS/MS and information-dependent analysis (IDA) nanoLC-MS/MS were used for random and targeted identification of disulfide-linked peptides. Finally, an example of electrospray-MS (ESI-MS) and ESI-MS/MS–based determination of disulfide-linked peptides is shown.
Introduction
In their efforts to determine protein structure, identification of the correct disulfide bridges has always been a challenge for structural biologists.1–7 This especially difficult task of identifying the disulfide bridges is particularly critical in creating restriction points in the 3D structure of a protein and differentiation between two possible crystal structures or between a correct crystal structure and an artifactual one.1,2,8–11 The correct disulfide formation also has significance for maintaining the physiological state of an organism or the onset of a pathological one. For example, misfolding of a secreted protein with scrambled disulfide bridges will prevent it from performing its cellular or extracellular (or physiological) function.3,8,10,12–25 In addition, a point mutation that would replace a cysteine (Cys) amino acid to a different one, thus preventing disulfide formation, would not only prevent the mutated protein from performing its function but also enable pathological protein-protein interactions and aggregations based on the free, unpaired Cys from the predicted disulfide bridge.1,3,26,27 Therefore, identification of disulfide bridges allows us to understand not only the structure of proteins but also their physiological and pathological states.1,3,26,27 Here we describe the current and past methods used to identify the disulfide bridges in proteins, with a focus on mass spectrometry (MS)–based methods and a particular emphasis on nanoliquid chromatography–tandem mass spectrometry (nanoLC-MS/MS) data-dependent analysis (DDA) and information-dependent analysis (IDA) and on electrospray-MS (ESI-MS) and ESI-MS/MS-based methods.
Analysis of Disulfide Bridges in Proteins by Non-MS-Based Methods
Historically, disulfide bridges were identified using 2D electrophoresis (2DE), with the first separation in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under nonreducing (NR) conditions followed by reduction and separation by SDS-PAGE under reducing (R) conditions in the second dimension.12,15,28 Both proteins and peptides that are disulfide linked can be identified. This method is also named two-diagonal electrophoresis.9,28 Using a similar principle, 2D chromatography (2DC, paper or silica) can also be used, especially for identification of peptide-based disulfide bridges. Blocking of free Cys residues with Elman’s reagent following 2DE or 2DC also allows for rapid identification of disulfide-linked Cys residues and their position within proteins. Another fast method for identification of disulfide bridges that is inexpensive, still in use, and effective is high-performance liquid chromatography (HPLC) analysis of disulfide-linked peptides or proteins that are separated under NR and then R conditions. The retention time could easily determine whether there is a disulfide bridge and, if so, where exactly within the protein sequence it is. Identification would then follow.
Analysis of Disulfide Bridges by MS-Based Methods
Over time, two main methods for analysis of disulfide bridges in proteins, both with advantages and disadvantages, have emerged. One is based on the use of MS with a matrix-assisted laser desorption ionization source (MALDI-MS), and the other one is based on the use of MS with an electrospray ionization source (ESI-MS), coupled with an HPLC (LC-MS/MS).
Analysis of Disulfide Bridges by MALDI-MS
This method is based on the identification of the shift in the mass of the disulfide-linked peptides or of peptides with intramolecular disulfide bridges.9,12,26,29–42 Ideally, the protein sample is digested with a protease (usually trypsin) under nonreducing and slightly acidic conditions, and then the peptide mixture is split in two: One half is co-crystalized with the matrix (e.g., HCCA), whereas the other half is reduced with dithiothreitol (DTT) and then co-crystalized with the matrix, followed by MALDI-MS analysis. Having the peptide mixture analyzed in both NR and R conditions allows one to identify the shift of the peaks that are responsible for disulfide bridge(s). In the simplest case scenario for an intermolecular bridge, one should identify (by MALDI-MS under NR conditions) one peak that corresponds to two peptides (P1 and P2) disulfide linked. In MALDI-MS analysis of the same protein sample, but under R conditions, the peak that corresponds to P1 and P2 disulfide linked (and observed in NR) should not be further observed in R conditions. Instead, two distinct peaks that correspond to the masses (singly charges) of the individual P1 and P2 peptides should be observed. Simply by comparing the two MALDI-MS spectra from NR and R, one could easily identify the disulfide-linked peptides. In the case of an intramolecular bridge, where only one peptide (e.g., P3) contains two disulfide-linked Cys residues, one should observe the oxidized P3 (P3 – 2Da) in NR and the reduced P3 in R. Therefore, a 2-Da increase in the P3 upon reduction should be observed.
For the more complicated disulfide bridges, the rationale for their identification should be the same as for the simple two-peptide intermolecular bridges or intramolecular bridges with more disulfides within the same peptide. For example, for three peptides that are disulfide linked, the disulfide should be calculated as follows: P1-SH plus P2-SH equals P1-S-S-P2 (a loss of 2 Da), whereas P1-SH plus HS-P2-SH plus P3-SH equals P1-S-S-P2-S-S-P3 (a loss of 4 Da). The charges due to the acidic conditions are extra (for a singly charged [1+] disulfide, add 1 Da, whereas for a [2+] disulfide, add 2 Da). For the intramolecular bridges, when there is one intramolecular bridge, the peptide has a loss of 2 Da, whereas for two bridges, there is a 4-Da loss for that peptide under NR conditions. More complex disulfide bridges, such as three or more disulfide-linked intermolecular peptides that also have intramolecular bridges themselves, are also possible.
One advantage for this method is that it can analyze disulfide-linked peptides with relatively high m/z, a feature not always available when using other methods. However, one drawback is that the disulfide-linked peptides may break apart during ionization or do not ionize at all, or they have to be confirmed by digestion using a different, sometimes complementary enzyme. A schematic of determining the disulfide-linked peptides is presented in Figure 1 .
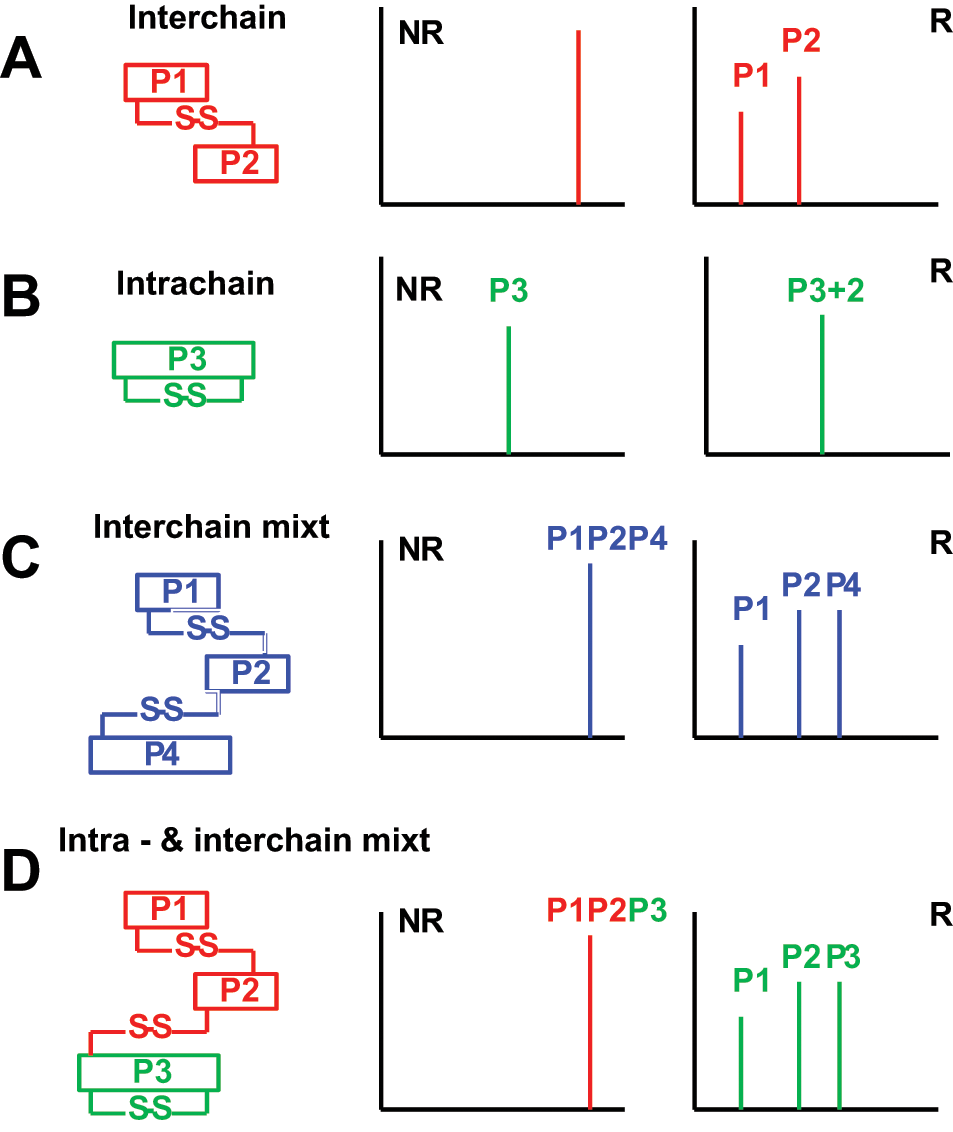
Principle of analysis of disulfide-linked peptides by matrix-assisted laser desorption ionization–mass spectrometry (MALDI-MS). The disulfide-linked peptides are intermolecular (
Analysis of Disulfide Bridges by LC-MS/MS Using DDA
There are two main possibilities of identifying the disulfide-linked peptides using LC-MS/MS: using DDA or IDA. When the sample is abundant, the disulfide-linked peptides may also be identified by using direct infusion using the syringe pump, bypassing the HPLC run. LC-MS/MS analysis of the disulfide-linked peptides using DDA involves analysis of the peptide mixture under both NR and R.9,12,19,26,29,43–48 In the first LC-MS/MS run, the peptide mixture contains peptides with their Cys in reduced form and alkylated by an alkylating agent such as iodoacetamide (IAA) or iodoacetic acid. Therefore, the Cys-containing peptides can be identified and used as an indicator of what Cys-containing peptides may be identified in a LC-MS/MS run as both single peptides or as part of a disulfide bridge. Usually, if a Cys-containing peptide with the Cys alkylated is not identified by a LC-MS/MS run, then it will most likely not be identified as part of a disulfide linkage. LC-MS/MS runs of the peptide mixture with Cys residues reduced but not alkylated may also help us in the identification of the fragmentation pattern of this peptide by MS/MS and of the product ions that can be produced. The second step in identification of the disulfide-linked peptides involves LC-MS/MS runs of intact disulfide-linked peptides. In this step, the Cys-containing peptides observed in the previous run (e.g., peptides P1 and P2) can be identified simply by calculating the m/z of the (2+), (3+), or (+4) ions and searching for them in the raw data. The advantage for this type of determination of the disulfide-linked peptides is that the samples do not require any special or particular preparation. The samples are prepared under NR and R conditions and then run by LC-MS/MS as a regular experiment. In fact, many researchers bypass the LC-MS/MS run of the reduced (and perhaps alkylated peptides) simply because one can calculate the predicted m/z of all the theoretical disulfide-linked peptides and search directly for these peptides in the raw data. Although more effective, this method works when a protein has few or very few Cys residues. However, if a protein has more than four theoretical disulfides, then this method becomes impractical. The drawback of this method is that it is time-consuming. Looking for the theoretical m/z of a peak that corresponds to a disulfide bridge in (2+), (3+), or (4+) charge state may take a very long time; however, it is not impossible. The full procedure for identification of disulfide-linked proteins by LC-MS/MS using DDA is shown in Figure 2 .
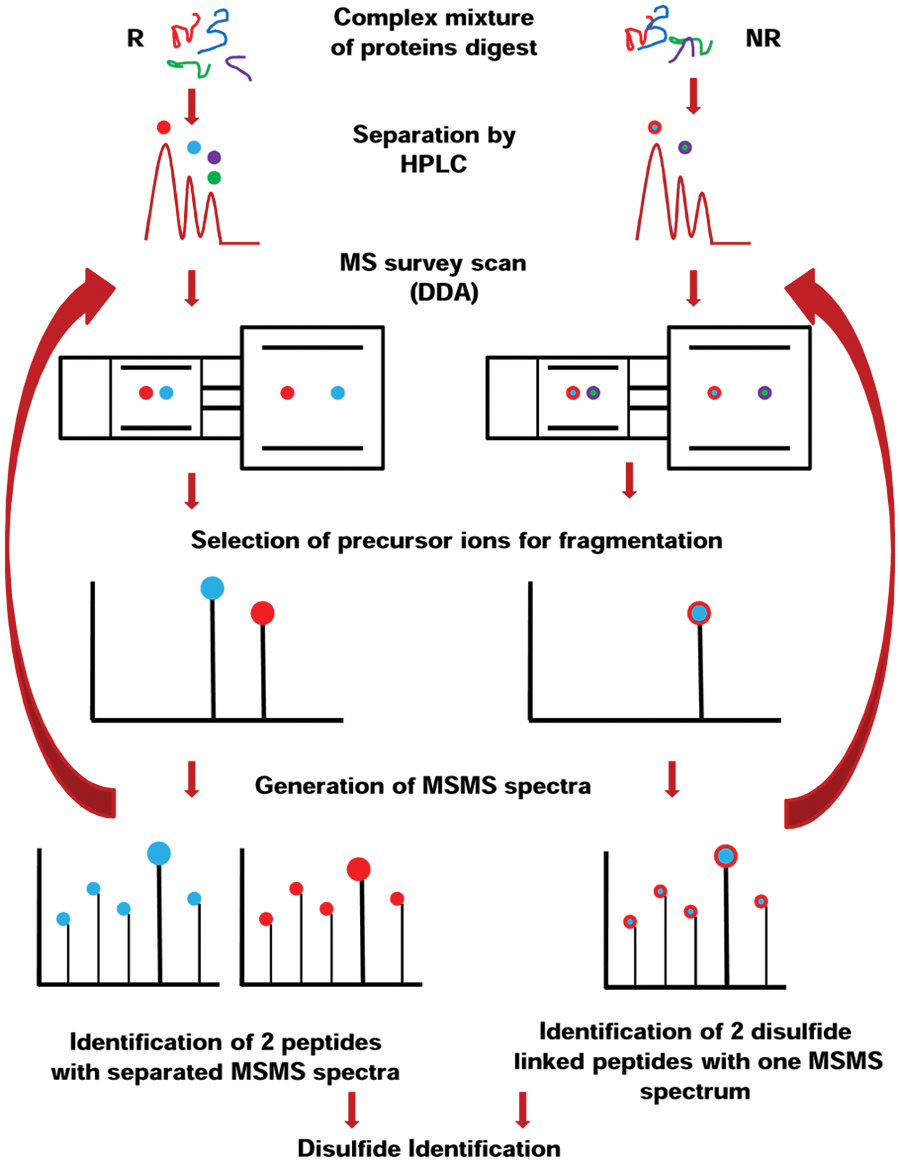
The principle of identification of disulfide-linked peptides using nanoliquid chromatography–tandem mass spectrometry (nanoLC-MS/MS) under nonreducing (NR) and reducing (R) conditions using data-dependent analysis (DDA). HPLC, high-performance liquid chromatography.
Analysis of Disulfide Bridges by LC-MS/MS Using IDA
Perhaps one of the best methods for identification of disulfide-linked peptides is by LC-MS/MS using IDA. This approach is somehow related to the DDA approach and derived from it. The principle is simple: If one calculates all theoretical possibilities for the disulfide connectivities in a protein, in (2+), (3+), or (4+) charge state, why not instruct the MS to look preferentially for some (or all) theoretical peaks that correspond to disulfide-linked peptides, instead of looking for one peak at the time? This instruction of the MS, named IDA, is based on including in the MS method a list with m/z for the peaks of interest. Depending on the preferences, the MS will then look for those peaks from the list preferentially—for example, do a regular DDA, but if a peak that corresponds to the a priori set criteria appears, then the peak is selected for fragmentation over other peaks with perhaps higher intensity (the peak is preferred). Alternatively, the MS method can be instructed to look only for particular peaks of interest (inclusion list only), where the peaks that are not on the list are not selected for fragmentation and are not fragmented.
The advantage for this method is that the MS can perform a regular DDA experiment when the inclusion list criteria are not met (in the preferred inclusion list mode) and switches to IDA when the inclusion list criteria are met. The outcome of this type of experiment is identification of one or more proteins (DDA when inclusion list criteria are not met) and their disulfide-linked peptides (IDA when inclusion list criteria are met). When the MS works in the inclusion list–only mode, the disulfide-linked proteins are identified at a higher rate, but the information about the protein(s) is missed. However, almost no researcher will perform an IDA LC-MS/MS experiment without knowing the identity of the protein(s) from the samples. The full procedure for identification of disulfide-linked proteins by LC-MS/MS using IDA is shown in Figure 3 .
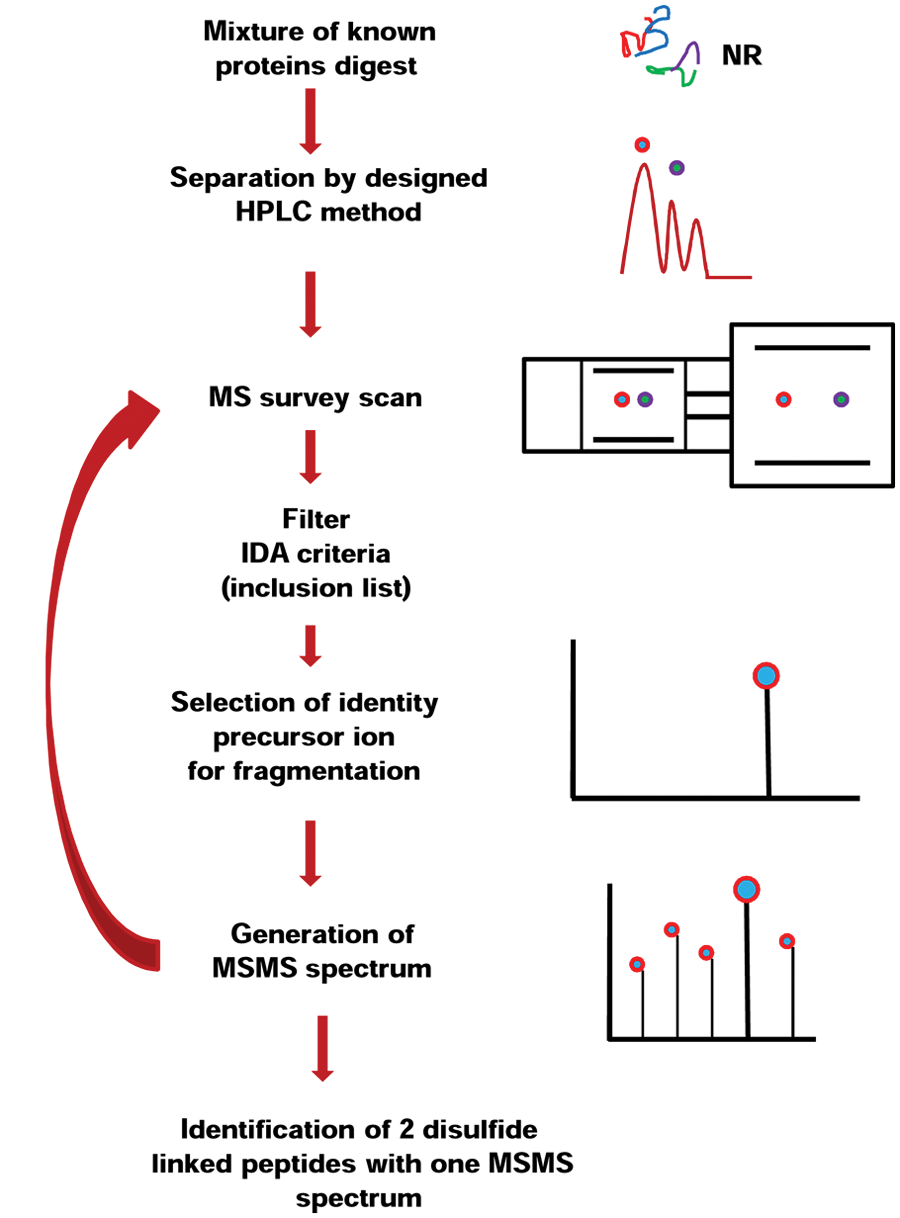
The principle of identification of disulfide-linked peptides using nanoliquid chromatography–tandem mass spectrometry (nanoLC-MS/MS) under nonreducing (NR) and reducing (R) conditions using information-dependent analysis (IDA). HPLC, high-performance liquid chromatography.
Determination of Disulfide Bridges Using ESI-MS and ESI-MS/MS
The existence of one or more disulfide bridges can simply be determined by ESI-MS, whereas identification of the exact location of the bridge is by ESI-MS/MS. By calculating the theoretical mass of a peptide (or a set of peptides) with a known sequence, one may determine whether that peptide (those peptides) form(s) disulfide bridges. Figure 4 shows such an example. Here, a peptide with a sequence PKLHEIRIEKANNLLYINC (calculated m/z of the singly charged peptide 2281.2485) was analyzed by ESI-MS and identified as a triply charged (3+) peptide with m/z of 761.08 ( Fig. 4A , B ) and (2+) peptide with m/z of 1141.12 ( Fig. 4A , C ). A third peak with m/z of 912.65 (5+) has a calculated m/z of 4559.25 for the singly charged peptide ( Fig. 4A , D ). Although the peaks with m/z of 761.08 (3+) and 1141.12 (2+) correspond to peptide PKLHEIRIEKANNLLYINC with Cys in reduced form, the peak with m/z of 912.65 (5+) corresponds to two peptides PKLHEIRIEKANNLLYINC disulfide linked at the Cys residue: m/z of [(2281.24 × 2 – 2) – 2] + 1 = 4559.48 (calculated theoretical singly charged peptide). Therefore, the peptide PKLHEIRIEKANNLLYINC is mainly detected as a monomeric peptide with the Cys reduced but also as a disulfide-linked homodimer.
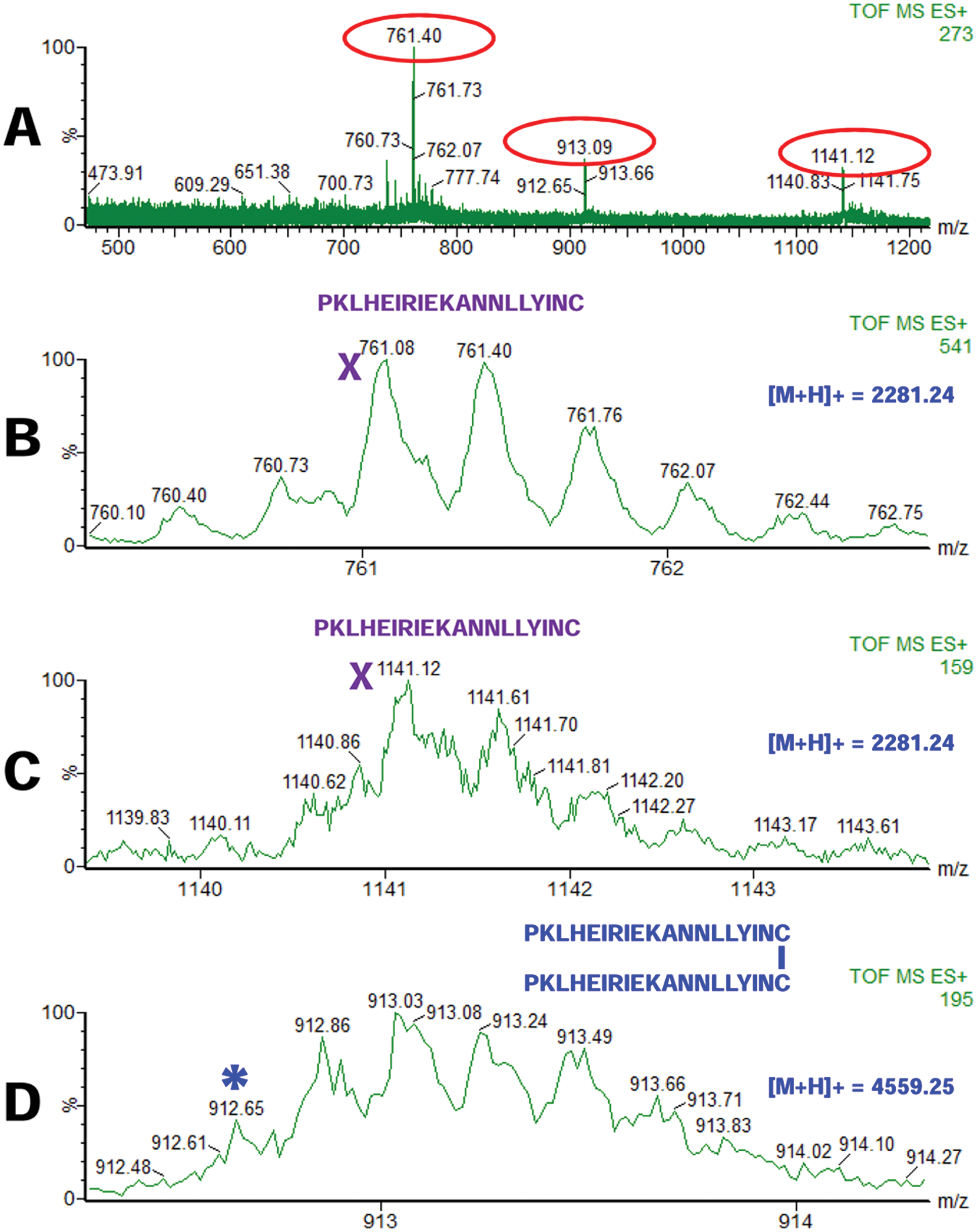
Electrospray–mass spectrometry (ESI-MS) of a peptide PKLHEIRIEKANNLLYINC in monomeric (with Cys residues reduced;
To demonstrate that the peaks observed in Figure 4 are indeed one and the same peptide but as a monomer or disulfide-linked dimer, we fragmented all three peaks observed in Figure 4A , with m/z of 761.08 (3+) ( Fig. 5A , D ), 1141.12 (2+) ( Fig. 5B , E ), and 912.65 (5+) ( Fig. 5C , F ). As observed, the MS/MS spectra of these peptides produced the same fragments and only b-type ions, but not y ions, suggesting that these three peaks indeed correspond to the same peptide.
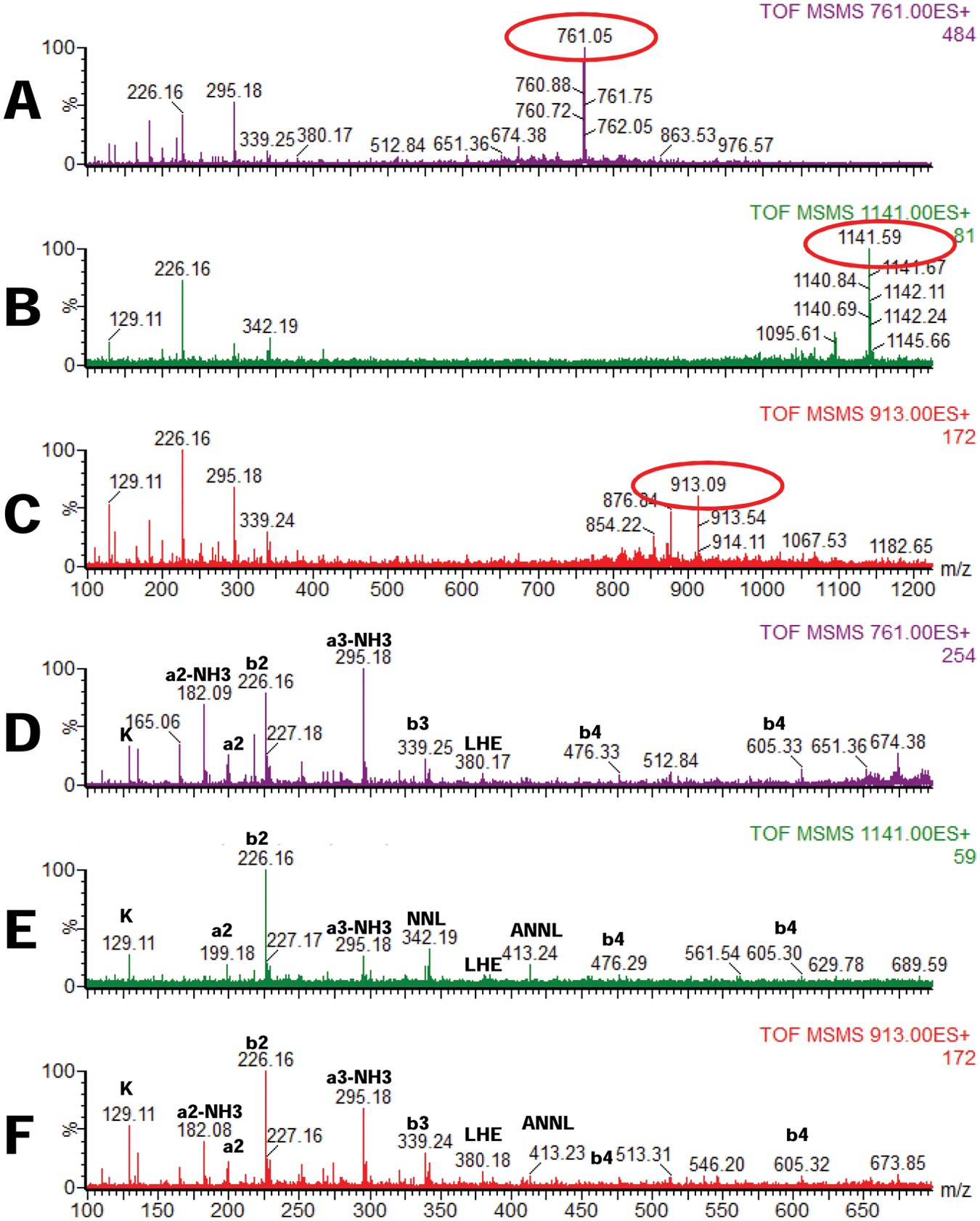
Electrospray–tandem mass spectrometry (ESI-MS/MS) of a peptide PKLHEIRIEKANNLLYINC in monomeric (with Cys residues reduced;
In some experiments, the isotopic envelopes of the 761.08 (3+) and 1141.12 (2+) peaks were not well resolved or not even very clear. Therefore, we suspected that the monomeric peptides form disulfide-linked homodimers. Since it was clear to us that the 912.68 peak is a disulfide-linked peptide, to further investigate the disulfide formation for this peptide, we also investigated whether the peptides that correspond to peaks with m/z of 761.08 (3+) and 1141.12 (2+) also form disulfides. We indeed found, in addition to the monomeric peptide with m/z of 761.08 (3+) and 1141.12 (2+), also the peaks that correspond to the disulfide-linked peptides with m/z of 760.73 (6+) and 1140.63 (4+). A summary of this experiment is shown in Figure 6 .
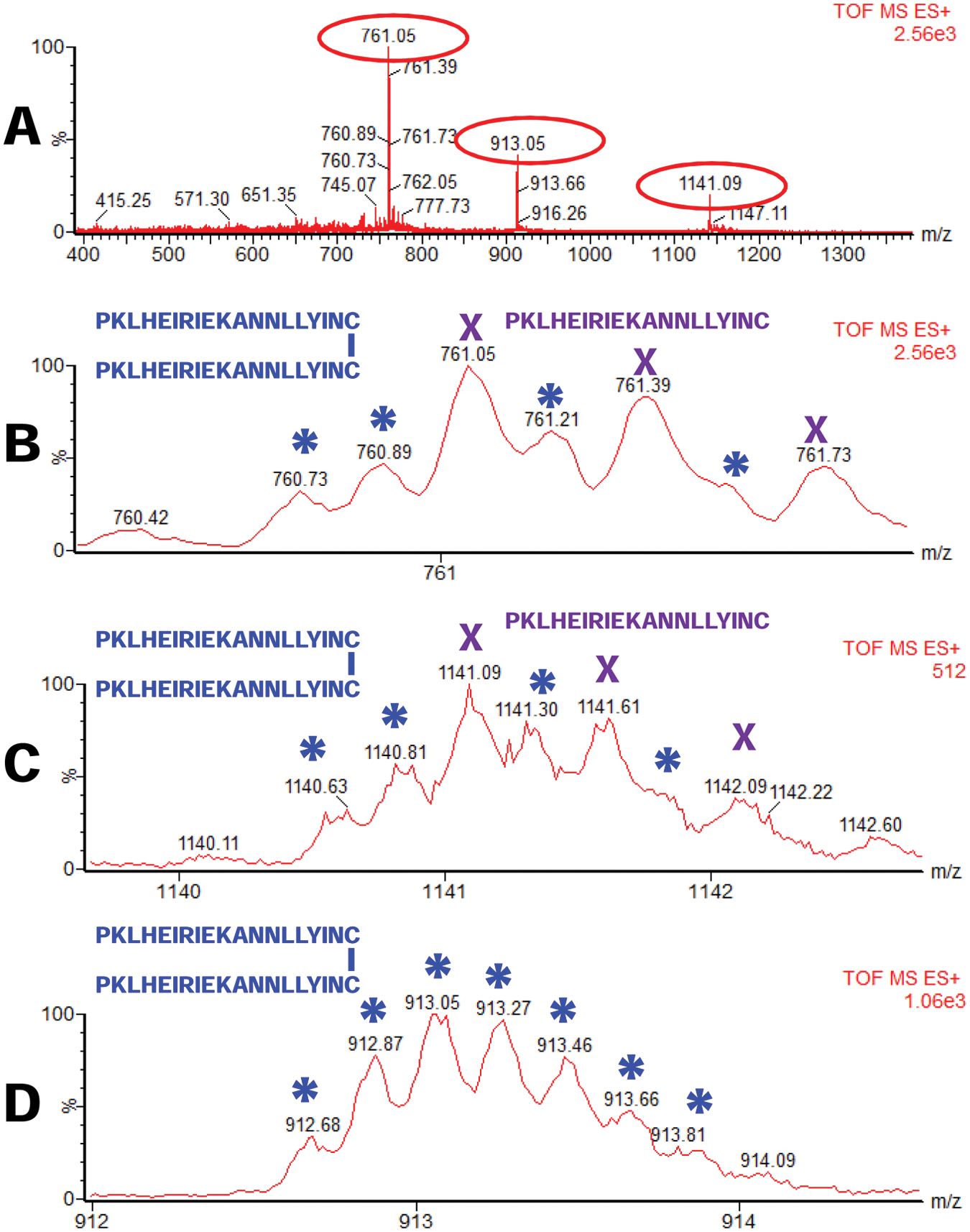
Electrospray–mass spectrometry (ESI-MS) of a peptide PKLHEIRIEKANNLLYINC in monomeric (with Cys residues reduced;
Interpretation of the MS/MS Spectra
Once the precursor ions that correspond to an intermolecular or intramolecular disulfide bridge have been identified by DDA or IDA and selected for fragmentation, the MS/MS of these precursor peaks must be sequenced and verified. The classical method is de novo sequencing. However, software-based de novo sequencing of disulfide bridges or software-only–based analysis is becoming more popular. One strong Web-based free available software tool is MS-bridge (from UCSF, http://prospector.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msbridgestandard), which helps one identify not only the disulfide-linked peptides but also cross-linked peptides with the common cross-linkers used in MS.
Current Challenges in Identification of the Disulfide Bridges
Although identification of one intramolecular disulfide bridge or two disulfide-linked peptides is relatively easy in theory, a lot of challenges make identification of the disulfide-linked peptides difficult. Among them, the biggest challenges are tri- or even tetrapeptides that are disulfide linked and the size of the peptides. If tri- or tetrapeptides can still be predicted and calculated and then identified using DDA or IDA analysis, the size of the peptides can still be a problem, simply because they usually have a high molecular mass (over 3000 Da), which makes it difficult to analyze (e.g., by quadrupole time-of-flight [QTOF] instruments, but not impossible to analyze by MALDI-MS). Another challenge for the identification of disulfide bridges are posttranslational modifications since most secreted proteins that form disulfide bridges are intensely posttranslationally modified. A particular difficult task is to analyze disulfide bridges in proteins that are posttranslationally modified by N-linked and/or O-linked oligosaccharides, mostly because it is very difficult (if not impossible) to predict the mass of the glycan and then to calculate the theoretical mass of the disulfide-linked peptides. Another challenge, and perhaps the most important one, is the sample preparation. Although mass spectrometry specialists are well aware of disulfide scrambling at alkaline pH, very few biologists are aware of this. Such experiments, performed at alkaline pH, are not only destined to fail, but the ones that succeed identify the wrong disulfide connectivities, which is perhaps the most dangerous piece of information to a structural biologist who tries to solve the structure of a protein, but this information conflicts with the experimentally detected disulfide bridges for that protein.
Although considerable progress has been made toward the identification of disulfide bridges in proteins, many proteins still do not yet have their disulfide connectivities determined. In addition, although disulfide bridges have been determined over time using a variety of methods, in the past few years, mass spectrometry has become the method of choice for most disulfide bridge determination. Furthermore, with the movement of the therapeutics from small molecular mass compounds to proteins and antibodies, determination of the disulfide bridges in these proteins has become a mandatory procedure for ensuring the correct linkage between Cys residues that will allow companies to produce and sell their product based on identification of the correctness. It is worth mentioning that this identification is more and more an automated MS-based method.
Footnotes
Acknowledgements
C.C.D. thanks Ms. Laura Mulderig and her colleagues (Waters Corporation) for their generous support in setting up the Proteomics Center within the Biochemistry & Proteomics Group at Clarkson University.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by Clarkson University (startup to C.C.D.), the Army Research Office through the Defense University Research Instrumentation Program (DURIP grant #W911NF-11-1-0304 to C.C.D.), and Keep A Breast Foundation (KEABF-375-35054).