
Abstract
Rosai–Dorfman disease (RDD) is usually characterized by painless bilateral cervical lymphadenopathy associated with fever and leukocytosis. Although the disease may occur outside lymphnodes, manifestation of skeletal system occurs in less than 8% of cases. In addition, presentation of this disease in a purely skeletal form without lymph nodes involvement is extremely uncommon. This case report describes a 17-year-old female with a pure skeletal presentation of RDD in the fibula. Trocar biopsy was performed, and immunohistochemical staining using S100 and CD68 was done to confirm the diagnosis.
Keywords
Introduction
Rosai–Dorfman disease (RDD) is a rare, nonmalignant disease that typically manifest as massive bilateral cervical lymphadenopathy and fever. 1 Other common manifestations are leukocytosis, elevated erythrocyte sedimentation rate (ESR), and hypergammaglobulinemia. 2 Although cervical lymphadenopathy is the most common manifestation, extranodal involvement of the disease has been reported in the literature. The most common extranodal sites include skin, nasal sinuses, and central nervous system. 3,4 Bone involvement occurs in less than 8% of cases. Furthermore, the manifestation of this disease in bone without associated lymphadenopathy is extremely uncommon, accounting for only 2% of all cases. 5 –7 Based on the study by Demicco et al., the bones involved include tibia, femur, clavicle, skull, maxilla, calcaneus, phalanx, metacarpal, and sacrum. 5
Case report
A 17-year-old female presented with a history of intermittent right knee pain for 5 months. She also had several episodes of undocumented low-grade fever that resolved with paracetamol. There was no accompanying history of upper respiratory tract infection, weight loss, and night sweats. Her past medical, personal, and family history were unremarkable. On physical examination, there were no signs of inflammation and deformity around the right knee. Palpation of the knee revealed no tenderness and palpable mass. Neurovascular assesment was also unremarkable. The range of motion of the right knee was full, and the patient was able to walk with normal gait pattern. Furthermore, palpation of inguinal, axilla, and cervical region revealed no lymphadenopathy on those areas. The rest of her physical examination was unremarkable.
Radiographic examination revealed a lytic lesion measuring that was eccentrically located on the anteromedial aspect of the fibular head with narrow zone of transition. There was no evidence of periosteal reaction, matrix production, or soft tissue mass. However, cortical erosion was observed on the anteromedial aspect of the fibular head (Figure 1). Skeletal survey showed no other lesion. On computed tomography (CT) scan, there was a well-defined osteolytic lesion in the proximal end of fibula without periosteal reaction, matrix formation, and soft tissue mass (Figure 2). Furthermore, plain magnetic resonance imaging (MRI) of the knee showed a bony expansile lesion along the anteromedial border of the fibular head exhibiting diffused T2-weighted hyperintense signal, measuring 2.1 × 2.7 × 3.6 cm3 (Figure 3).
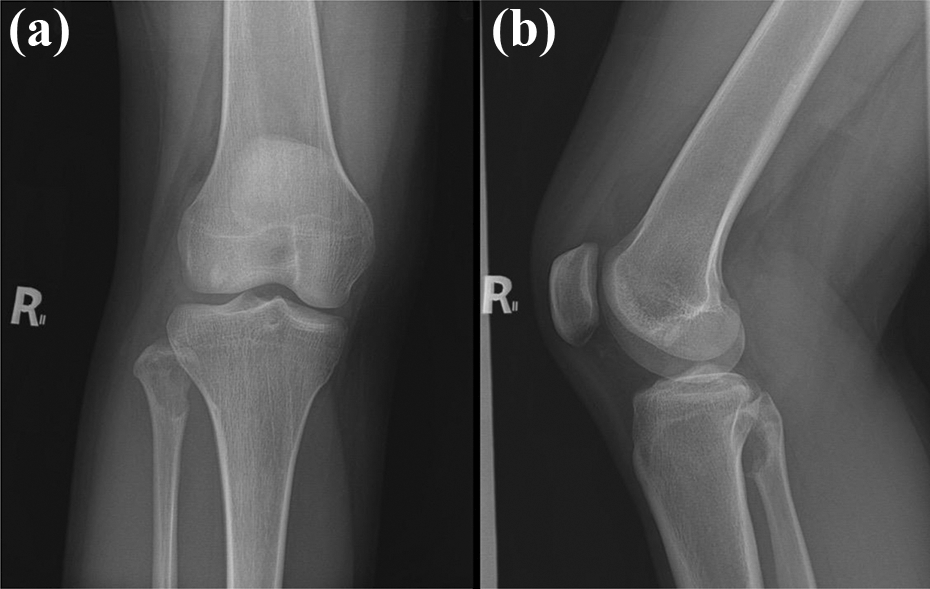
(a) Anteroposterior and (b) lateral radiographs of the right knee show a lytic lesion that is eccentrically located on the anteromedial fibular head with cortical erosion and narrow zone of transition. There is no evidence of periosteal reaction, matrix production, and soft tissue mass.
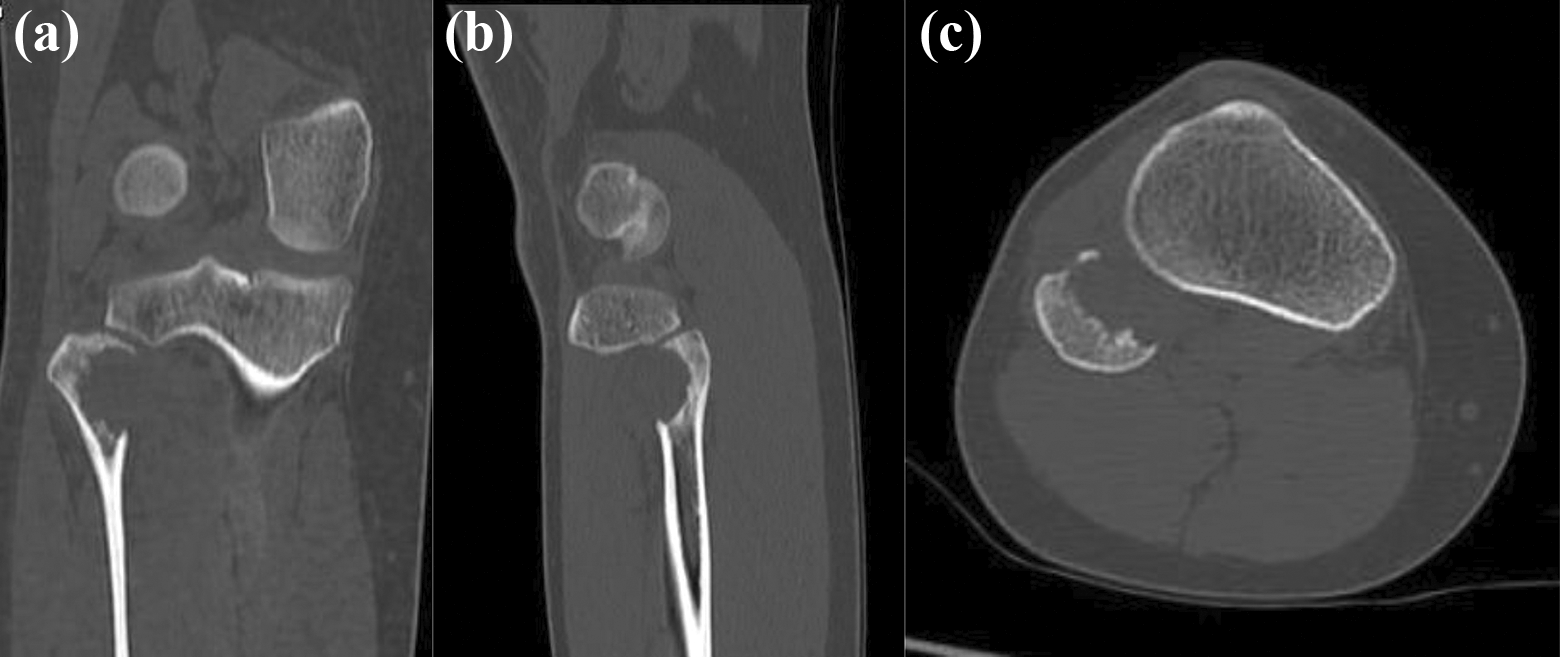
(a) Coronal, (b) sagittal, and (c) axial CT scan of the right knee through the lesion show a well-defined osteolytic lesion in the proximal end of fibula without periosteal reaction, matrix formation, and soft tissue mass.
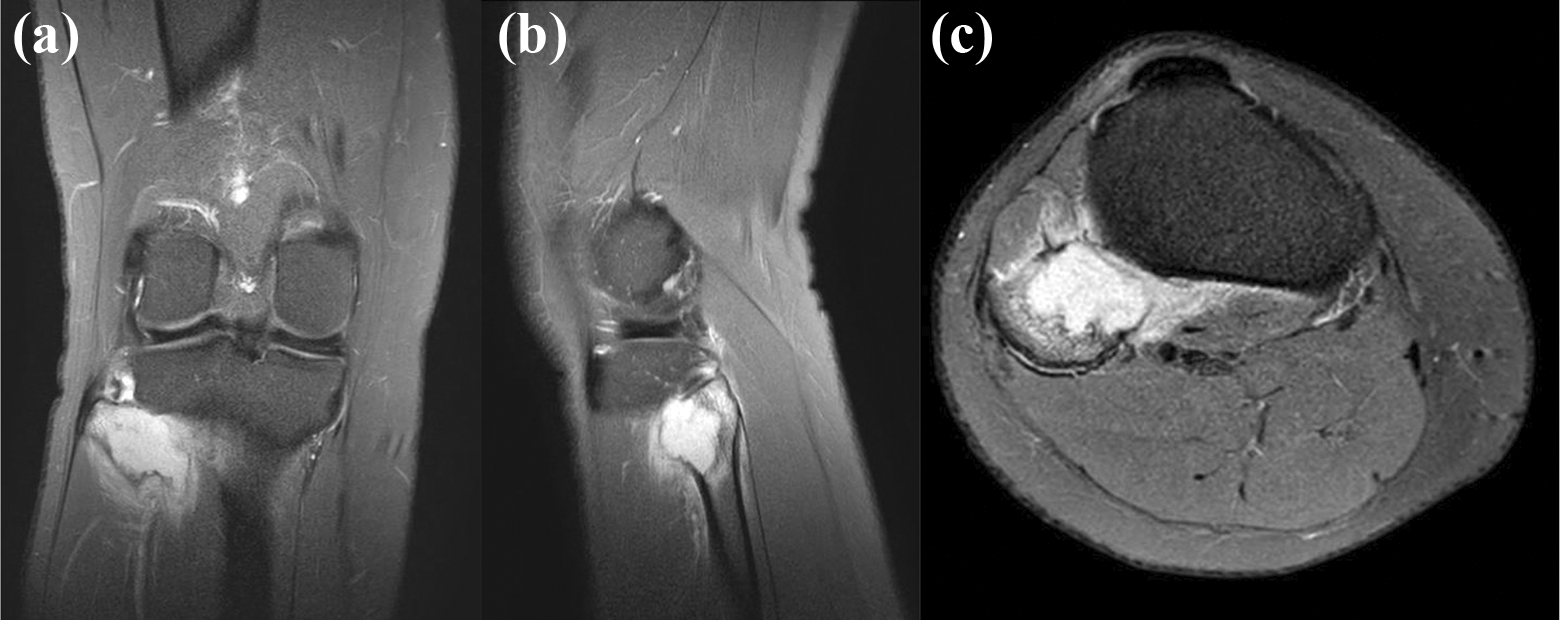
(a) Coronal, (b) sagittal, and (c) axial cut of T2-weighted MRI of the right knee through the lesion show a bony expansile lesion exhibiting diffused hyperintense signal along the anteromedial border of the fibular head.
Hematological investigation revealed normal findings. ESR was not elevated, and C-reactive protein was nonreactive. Lactate dehydrogenase, alkaline phosphatase, and total serum calcium are also within the normal range. At that time, the differential diagnoses were desmoplastic fibroma, giant cell tumor, and non-ossifying fibroma.
Trocar biopsy of the lesion was then performed under local anesthesia. The specimen was stained with hematoxylin and eosin. On microscopic examination, the presence of diffusely distributed histiocytes in the background of lymphocytes, plasma cells, and neutrophils was revealed. The histiocytes demonstrated emperipolesis of lymphocytes and neutrophils (Figure 4). At this point, the histopathological diagnosis was nonspecific acute and chronic inflammation with a consideration of sinus histiocytosis. Therefore, immunohistochemical staining with S100 and CD68 was performed. Both S100 and CD68 immunohistochemical staining were diffusely positive among the cells of interest, which indicate a lesion of histiocytic origin (Figures 5 and 6). Based on this characteristic cytomorphology, a diagnosis of RDD was made.
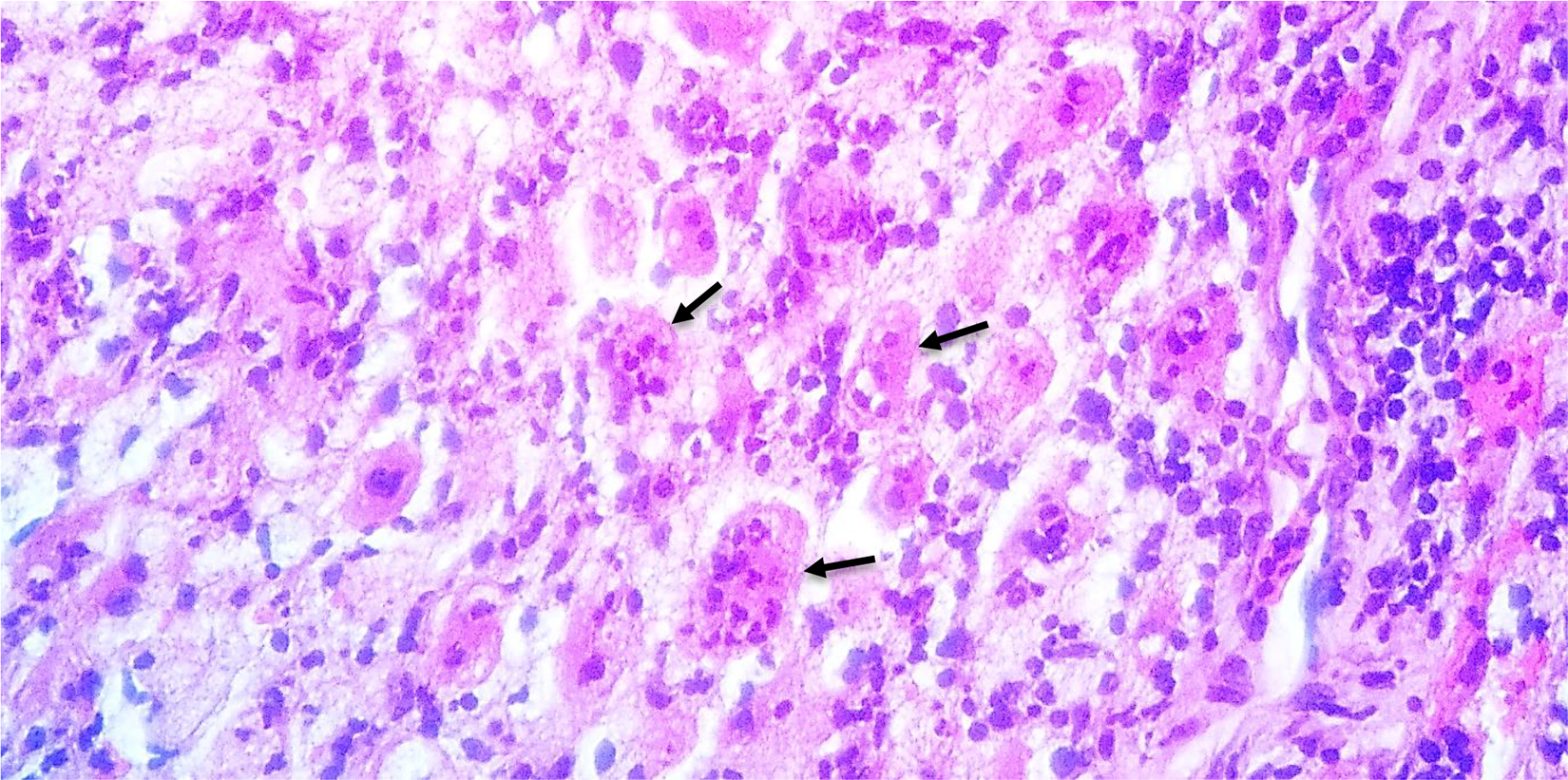
Histopathological examination shows the presence of diffusely distributed histiocytes in the background of lymphocytes, plasma cells, and neutrophils. The histiocytes demonstrated emperipolesis of lymphocyte and neutrophils (arrow). H&E: ×400. H&E: hematoxylin and eosin.
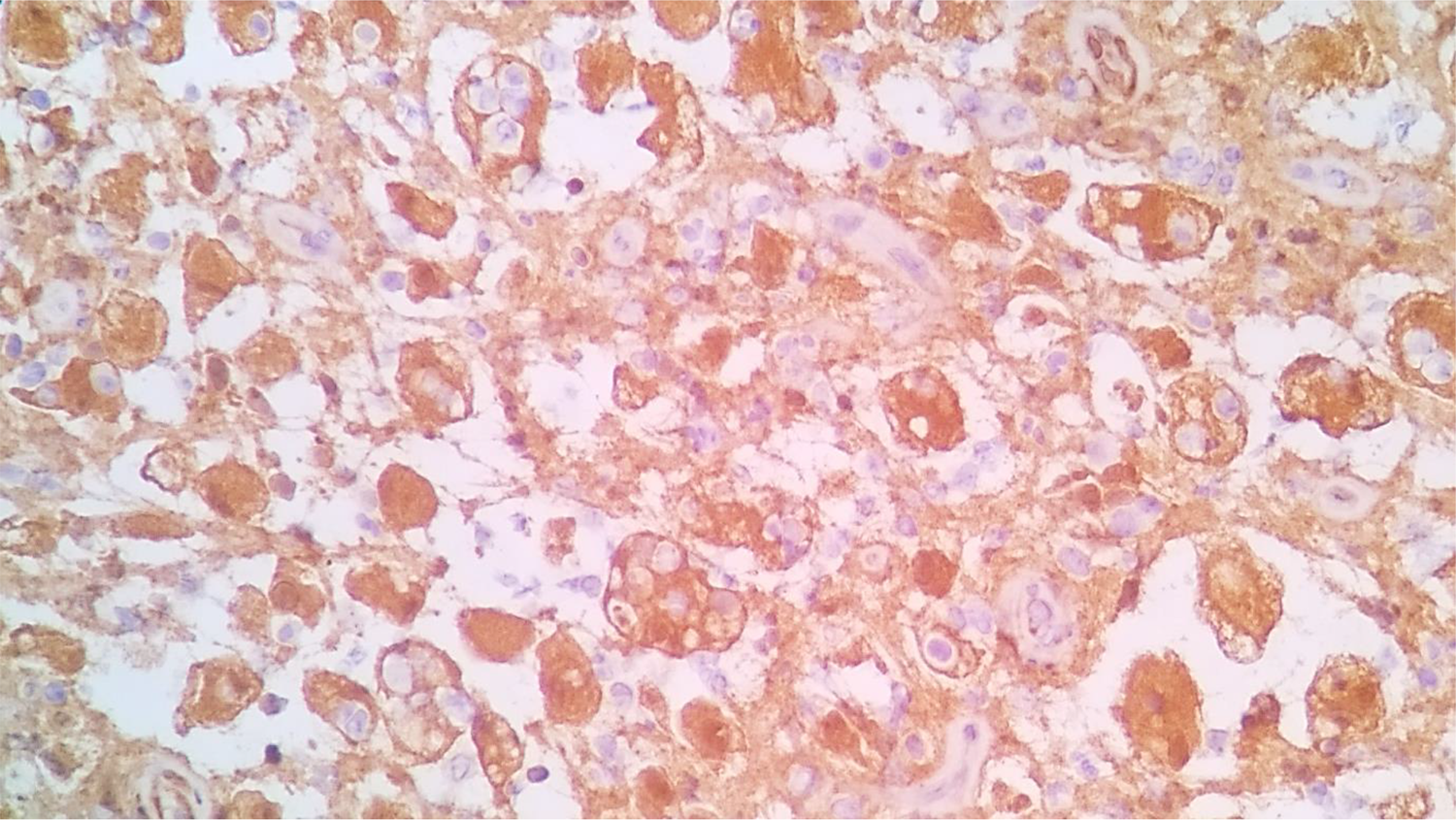
Immunohistochemical staining shows positive for S100.
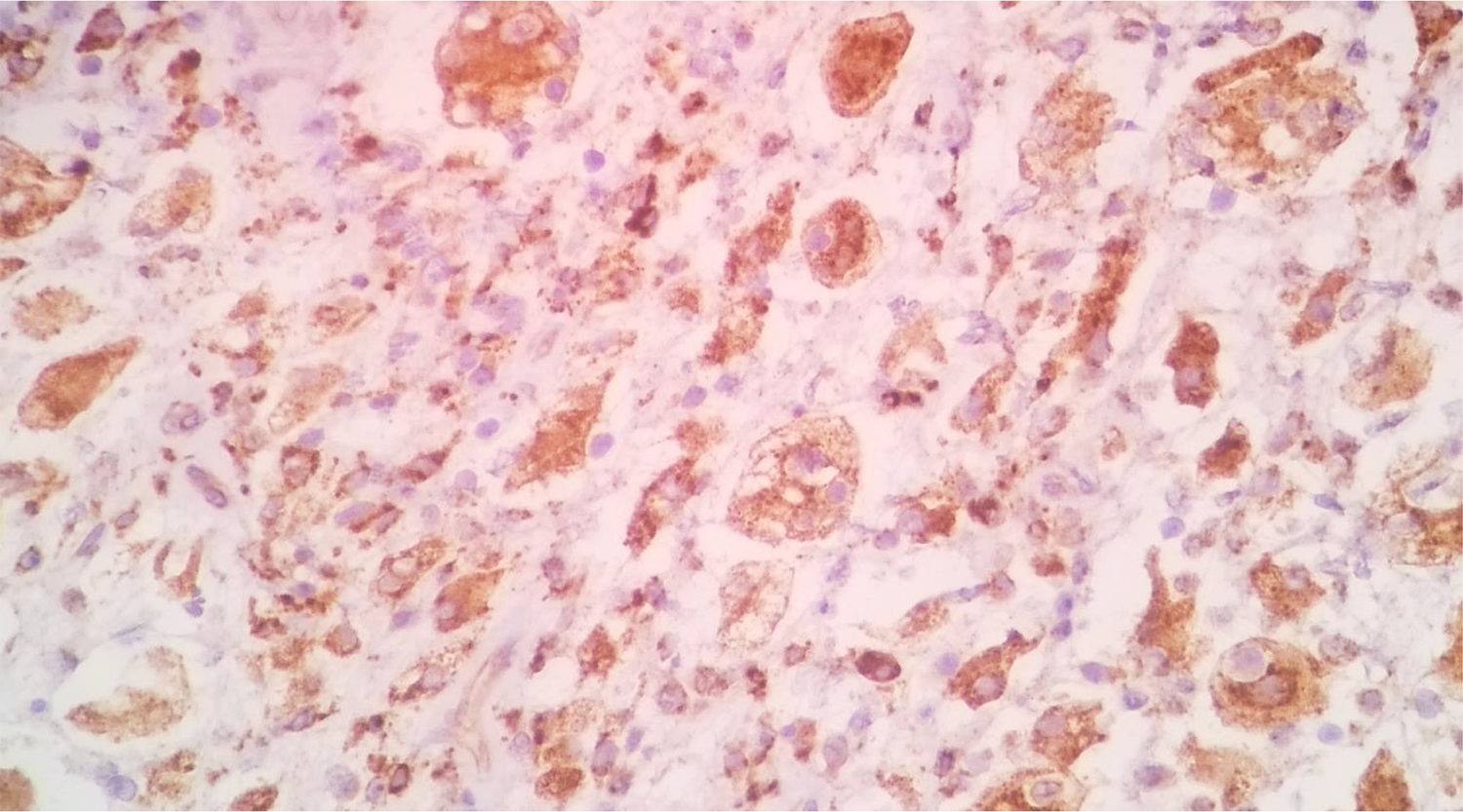
Immunohistochemical staining shows positive for CD68
Contrast MRI of chest and abdomen confirmed pure extranodal manifestation of the disease (Figures 7 and 8). Serum protein electophoresis revealed hypoalbuminemia and hypergammaglobulinemia (Figure 9 and Table 1).
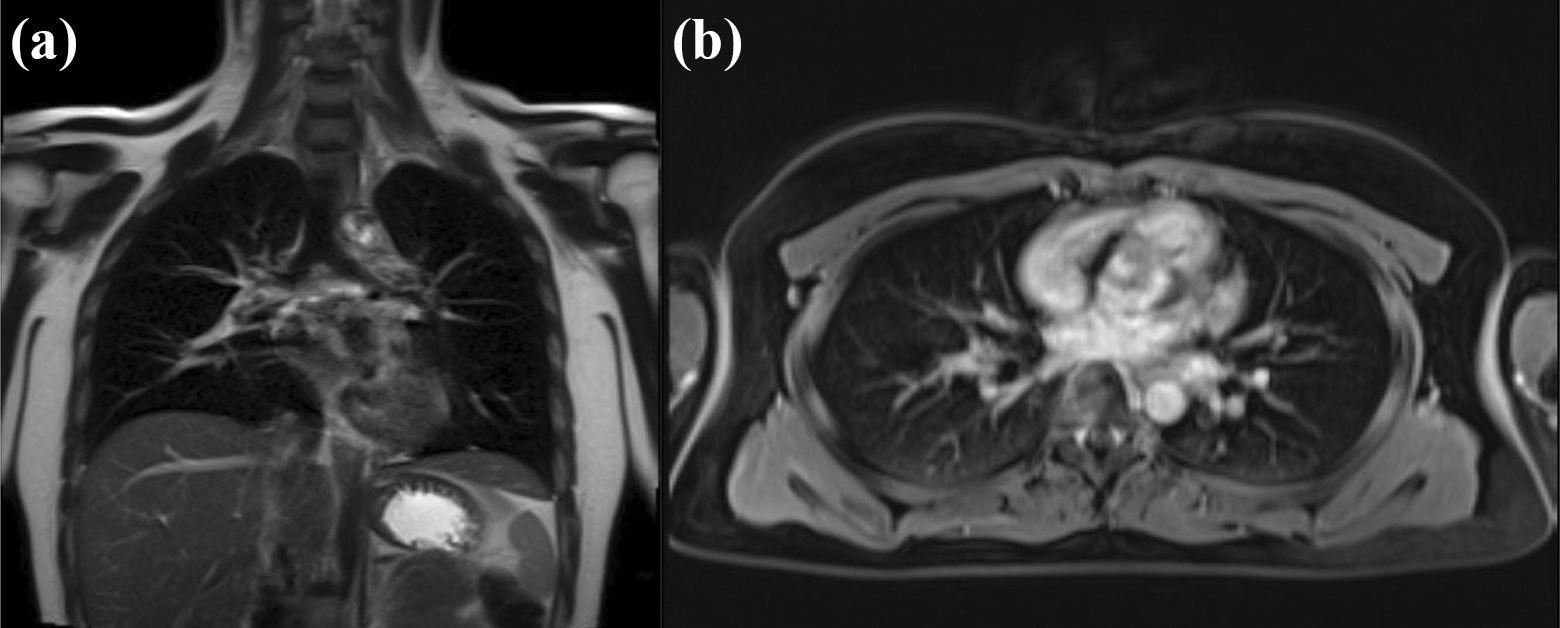
(a) Coronal and (b) axial cut of chest MRI with contrast show neither mass nor lymphadenopathy. MRI: magnetic resonance imaging.
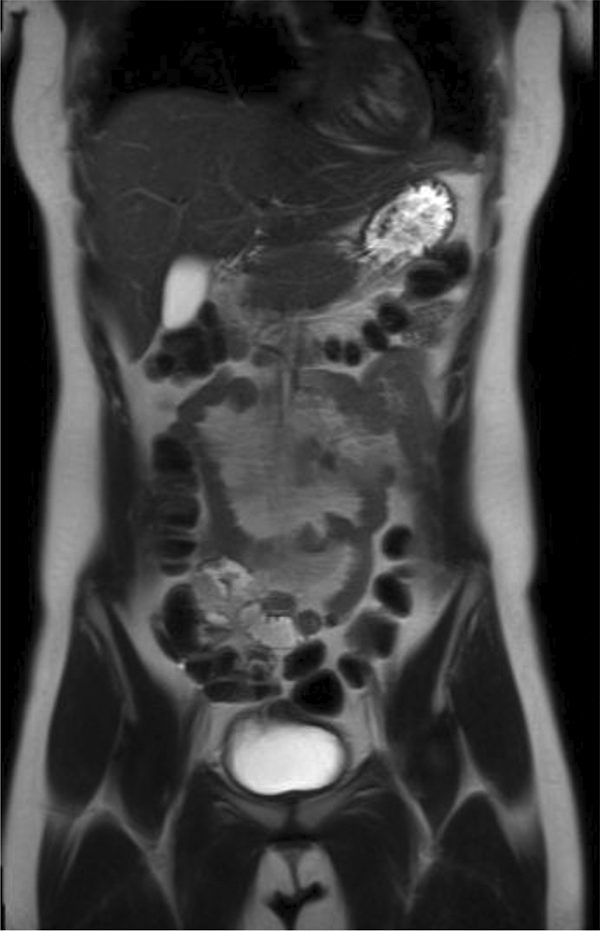
MRI of abdomen with contrast shows neither mass nor lymphanedopathy. MRI: magnetic resonance imaging.
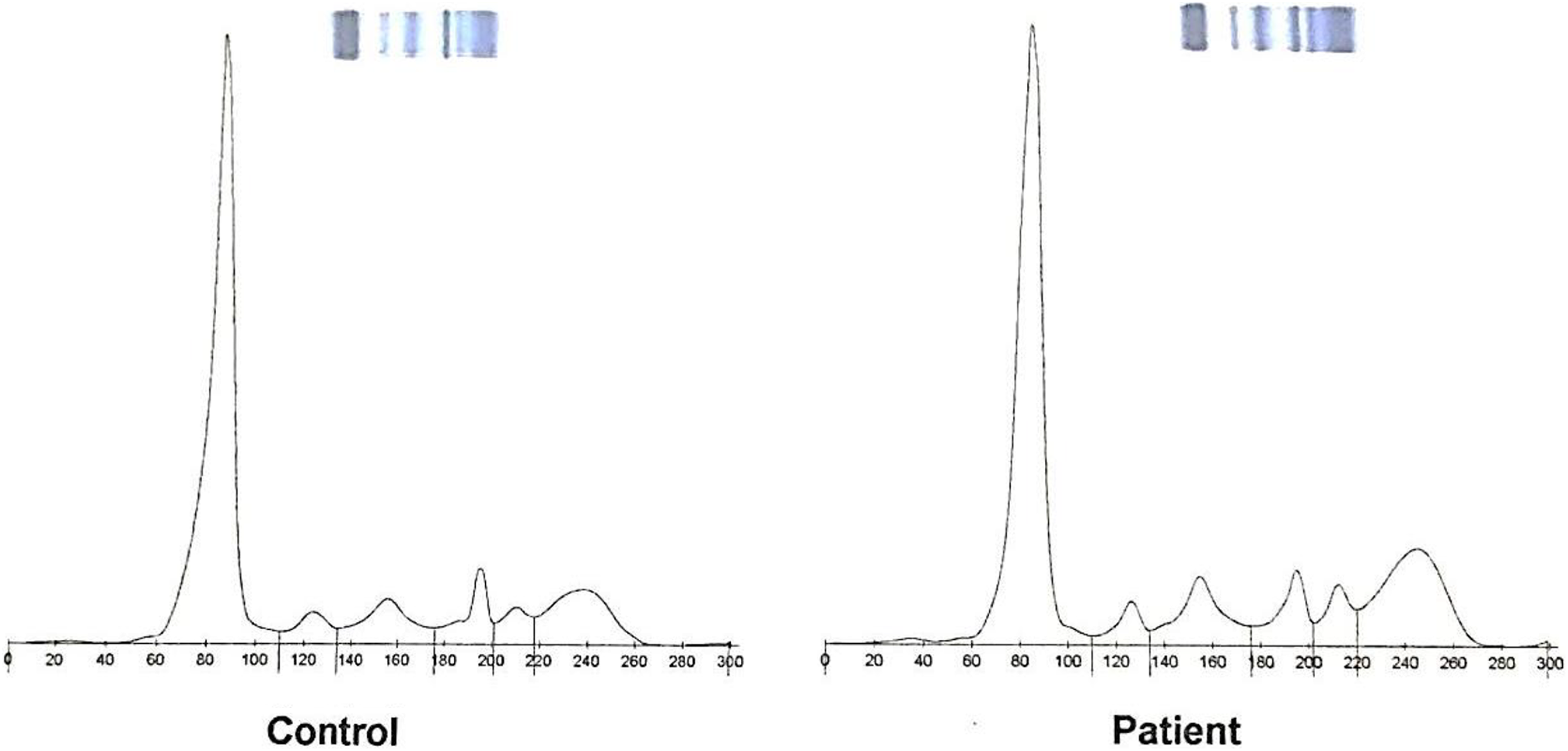
Serum protein electrophoresis graph of the patient shows an increase in gamma zone.
Serum protein electrophoresis profile of the patient shows hypoalbuminemia and hypergammaglobulinemia.
Since the patient had intermittent mild pain without sign of compression of peroneal nerve, she was only given NSAID as needed for pain and advised close observation at 3- to 6-month intervals with serial laboratory studies (complete blood counts and ESR) and radiographs.
During the 3-year follow-up, the patient had no complaint on her right knee and her physical examination was unremarkable. Laboratory investigation showed normal results. The latest radiographic examination and MRI revealed there was decrease in the size of the lesion on the fibular head. On T2-weighted MRI, the hyperintense bone lesion at the fibular head had the measurement of 1.8 × 2.5 × 3.2 cm3. There is no significant interval change in the extent of the adjacent soft tissue edema (Figure 10).
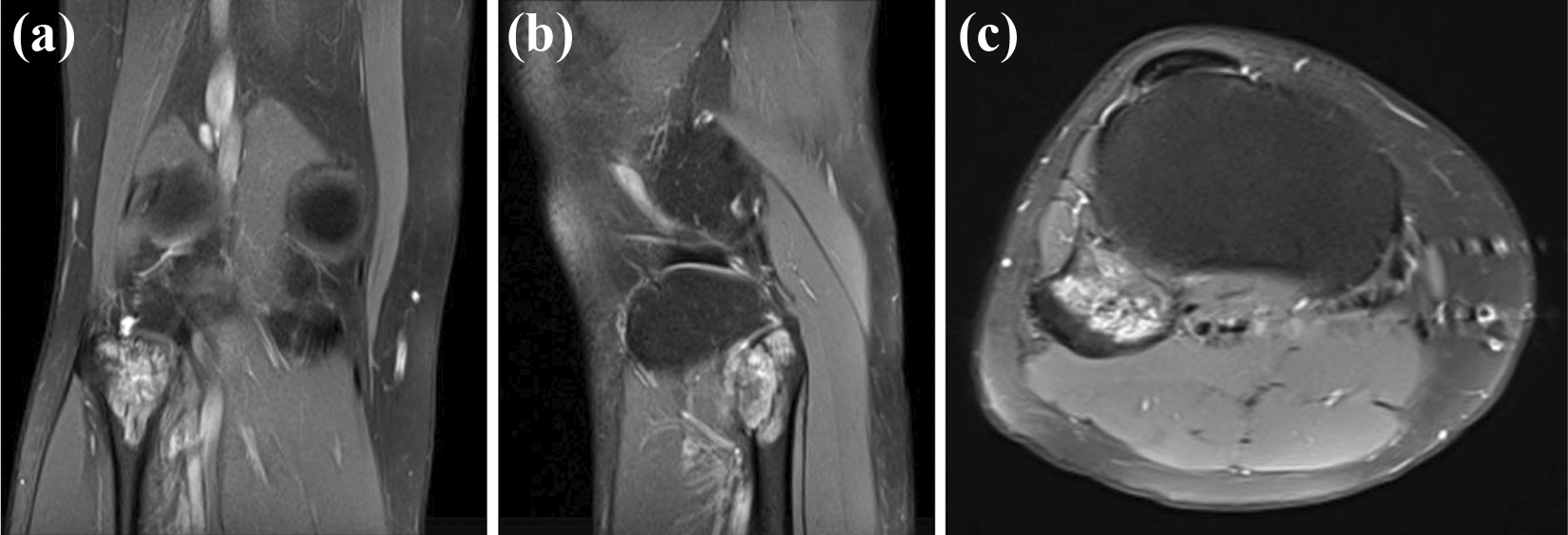
(a) Coronal, (b) sagittal, and (c) axial cut of T2-weighted MRI of the right knee through the lesion at 3-year follow up show slight interval regression of the lesion compared to the previous MRI. MRI: magnetic resonance imaging.
Discussion
RDD was first described by Juan Rosai and Ronald Dorfman as sinus histiocytosis with massive lymphadenopathy in 1969. This is a rare hystiocytic disorder which involves the overproduction of non-Langerhans sinus hystiocytes and accumulation of these cells most often in the lymph nodes. 1 It is most commonly seen in children and young patients in their second and third decades of life. A slight male predominance and general predilection for individuals of African descent have been described in the literature. 3,4,7
The etiology and pathogenesis of RDD are still unknown. It has been postulated that viral agents such as human herpes virus-6 and Epstein–Barr virus play a role in the pathogenesis of this disease. The abnormal reaction of the hematolymphoid system to infection leads to the initiation of monocyte colony-stimulating factor that mediates proliferation of histiocytes. Histiocytes that phagocytose numerous intact lymphocytes, termed emperipolesis, is the distinctive characteristic finding in RDD. 3,4,8
Classically, most patients present with massive bilateral cervical lymphadenopathy and fever. They also have leukocytosis, elevated ESR, and hypergammaglobulinemia. 1 –3 Patients may have normochromic/normocytic anemia or autoimmune hemolytic anemia. 3,9 Except for hypergammaglobulinemia, the aformentioned features are not present in this case.
Based on the histological findings, emperipolesis, which is the classical histological appearance of the disease, is present on this case. Moreover, positive finding of immunohistochemical staining with both CD68 and S100 is typical for RDD. 3,4
RDD of bone is a very rare condition. Manifestation of this condition in the skeletal system without lymphadenopathy is extremely uncommon, accounting for only 2% of all cases. 5 –7 Demicco et al. described 15 cases of primary RDD of bone which involved the tibia, femur, clavicle, skull, maxilla, calcaneus, phalanx, metacarpal, and sacrum. 5 Bone lesions appear usually in metaphysis or diaphysis of bone as osteolytic or mixed lytic sclerotic lesion with narrow zone of transition. 10,11 A thorough review of the literature had no yield describing RDD with a sole presentation affecting the fibula.
RDD is a self-limiting condition so that surgical resection or medical treatment is not required. It has been noted that greater than 70% of cases have spontaneous regression within 3 to 9 months. Furthermore, no malignant transformation has been reported in the literature. Hence, in most cases, the prognosis of the disease is excellent. 3,4,7,9
Active management of disease is only required for symptomatic patients. In patients with cervical lymphadenopathy resulting in a mechanical blocking of the airway, surgical management can be done to secure airway patency. For extranodal RDD, surgery is also an option if patients are symptomatic but with localized form of the disease. 3,4 Several studies have documented good overall outcome with curettage or surgical resection. The use of a bone graft to fill up the defects after curettage has also been documented in the literature. 12,13 Radiotherapy with doses of 20–50 Gy can also be used as adjunctive therapy. However, no standard radiation guidelines have been established due to the rarity of this disease. 3,5,7
Symptomatic patients who have systemic involvement may benefit from chemotherapy. The first line of chemotherapy that produces a response in both typical and extranodal RDD is the administration of steroids. Other chemotherapy agents that have been used are vinca alkaloids, anthracyclines, alkylating agents, methotrexate, cladribine, clofarabine, imatinib, retinoids, rituximab, and interferon. No clinical trial has been performed to compare the efficacy of these chemotherapeutic agents because, as previously mentioned, the disease is rare. 3,4,7,9 Since the patient has a localized disease with minimal symptom, she was only advised close observation at regular intervals.
Conclusion
RDD which presents in a purely skeletal form without lymph nodes involvement is extremely rare. Diagnosis can be made from biopsy which is confirmed with immunohistochemical staining. No active management is required for localized disease without symptoms. This case report contributes to the limited literature that documents the disease presentation, management, and prognosis of RDD.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.