
Abstract
Rosai–Dorfman disease (RDD) is a rare benign histiocytic proliferative disorder that classically involves lymph nodes. It often requires histopathological and immunohistochemical confirmation as it can often mimic malignancy on clinical examination and imaging. Treatment recommendations for this disease are widespread, with no clear universal approach. We present a 59-year-old otherwise healthy female with a rapidly growing and therapeutically challenging red-brown nodular plaque on the left cheek, which was consistent with an uncommon plaque-type variant of cutaneous RDD on histopathological evaluation. This case emphasizes the clinical variability of RDD and underscores the importance of histopathological evaluation to help clarify diagnosis, investigations and guide management for patients presenting with unusual clinical variants.
Introduction
Rosai–Dorfman disease (RDD), also called sinus histiocytosis with massive lymphadenopathy, is a rare form of non-Langerhans cell histiocytosis. 1 This benign histiocytic proliferative disorder classically involves lymph nodes and is diagnosed histopathologically and immunohistochemically as it can mimic malignancy on clinical examination and imaging. 2 Extra-nodal variants are noted in up to 43% of cases, with 10% of RDD patients exhibiting accompanying cutaneous lesions, and 3% presenting with cutaneous-only RDD (CRDD). 3 While the underlying cause of RDD remains poorly understood, prognosis is typically favorable, with a range of treatments reported from the limited number of cases. 2 We present the case of a 59-year-old female with a rapidly growing red-brown nodular plaque on the left cheek, consistent with an uncommon plaque-type variant of CRDD on histopathological evaluation.
Case report
A 59-year-old female was referred for assessment of a left cheek lesion. It began as a small papule which was increasing in size. It was not responsive to topical tacrolimus 0.1% ointment and had progressed over 6 months to form a 5 cm × 7 cm, well-demarcated erythematous to red-brown, indurated, bosselated plaque, with superficial telangiectasia and minimal epidermal changes (Figure 1(a)). It was firm, pruritic, and tender to touch, without any exudate or bloody discharge. The patient did not have any lymphadenopathy or organomegaly and denied any constitutional symptoms.
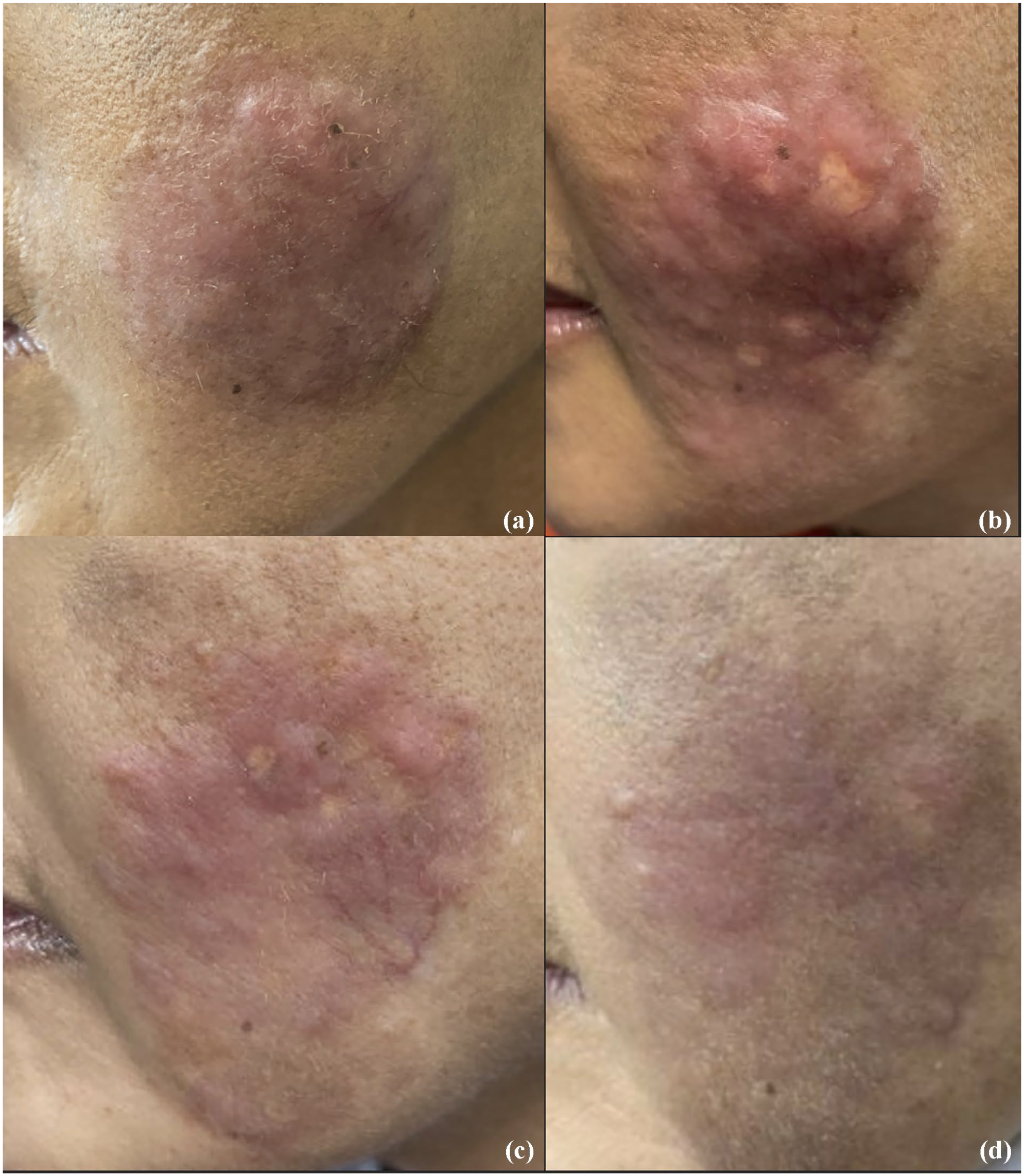
(a) Well-demarcated red-brown, indurated, bosselated plaque, with overlying telangiectasia and minimal epidermal changes on the left cheek, prior to initiation of treatment. (b) Update after 2 months and three intralesional corticosteroid injections (10–20 mg/mL) with evidence of new atrophic macules at injection sites. (c) Flattened plaque with less prominent nodularity 14 months after the initial consultation, following a total of six intralesional corticosteroid injections and initiation of oral corticosteroids. (d) Interval improvement with decreased telangiectasias, islands of normal skin and patchy nodularity at 22 month follow-up, after the completion of a tapering course of oral prednisone and five intralesional 0.5 mL injections of 5-fluorouracil 50 mg/mL.
The diagnostic differential included pseudolymphoma, lymphoma, tumid lupus, granuloma faciale, sarcoidosis, and infectious causes. An incisional wedge biopsy was sent for tissue histology, as well as bacterial, mycobacterial, and fungal cultures. The biopsy showed a lymphohistiocytic infiltrate positive for histiocyte marker CD163 and S100, as well as emperipolesis (Figure 2(a) and (b)). Tissue cultures were negative and the diagnosis of RDD was made.

(a) Hematoxylin-eosin staining shows lymphohistiocytic infiltrate, plasma cells, and histiocytes that are large with pale, foamy cytoplasm (original magnification 40×). (b) Numerous cells staining positively for S-100, with a green circle to highlight the evidence of emperipolesis (original magnification 40×).
Investigations were performed to rule out systemic disease and underlying malignancy. Her chest X-ray was unremarkable with no consolidations or lymphadenopathy. She had known longstanding mild normocytic anemia (Hgb 101 g/L; N: 115–155 g/L), and mild thrombocytosis (platelets: 462 × 109/L, N: 130–380 × 109/L). The immunofixation electrophoresis and urine albumin to creatinine ratio were unremarkable.
Hematology was consulted and conducted further investigations including an anemia work-up, liver function tests, extended electrolytes, immunoglobulins, Ebstein-Barr virus, Cytomegalovirus, and Human immunodeficiency virus serologies, anti-nuclear antibodies, rheumatoid factor, as well as CT chest, abdomen, pelvis, and head. All testing was negative apart from indeterminate bilateral pulmonary upper lobe opacities on CT chest, elevated vitamin B12 (1368 pmol/L; N: 201–731 pmol/L), and mild hypercalcemia (2.63 mmol/L; N: 2.24 2.58 mmol/L).
Treatment options were discussed, including observation, intralesional triamcinolone acetonide injections, systemic corticosteroids, systemic retinoids, radiotherapy, and surgical excision. After six sessions of injections of triamcinolone 10–20 mg/ml injected every 6 weeks, there was marginal improvement in areas treated, but progression was noted at the periphery and areas between injection sites remained indurated (Figure 1(b)). She then had a tapering course of oral prednisone starting at 40 mg daily and held the dose at 20 mg daily for 4 months before tapering off (Figure 1(c)). There was more improvement, but a steroid sparing option was desired. During the latter 2 months of the prednisone course, she received five 0.3–0.5 mL intralesional 5-fluorouracil (5-FU) 50 mg/mL injections. There were no significant side effects associated with 5-FU injections. She was also started on oral methotrexate, initially 10 mg weekly and increased 2 months later to 15 mg weekly, which has been well tolerated. Significant interval improvement was noted in the nodular plaque, which had decreased in size to 4.4 cm × 3.2 cm, with less telangiectasias and notably improved nodularity and induration (Figure 1(d)).
Discussion
RDD is a rare form of non-Langerhans cell histiocytosis, with an estimated prevalence of 1 in 200,000 and incidence of approximately 100 new cases per year reported in the United States. 1 Histologically, there is positive immunohistochemical staining for the more specific S100 marker. RDD classically presents with diffuse lymphadenopathy and can have constitutional symptoms. 2 Extra-nodal manifestations occur in 40% of cases including cutaneous, and rarely intracranial or ophthalmic involvement. 2 The etiology of RDD remains poorly understood. An increase in IgG4-positive plasma cells in RDD has been suggested, but not proven. 4 It has also been associated with HHV-6 reactivation, lymphoma, polyclonal hypergammaglobulinemia, and autoimmune diseases.3,5
CRDD accounts for approximately 3% of patients with RDD, usually presenting with small, red-brown, papules on the cheeks, and is increasingly recognized as a separate entity. 2 The patient in this case presented with CRDD and had a solitary, nodular plaque variant restricted to the cheek. As of 2016, only 20 cases have described cutaneous involvement limited to the cheek 6 and in a case series of 25 patients with RDD in China, only 12.8% were plaque-type. 7
Recently published consensus recommendations for the clinical management of RDD have suggested a wide spectrum of treatment options including observation, excision, radiotherapy, systemic corticosteroids, thalidomide, lenalidomide, and rituximab. 8 While a previous literature review supported observation as a management approach given spontaneous resolution in up to 50% of RDD cases, 9 a 2020 study found that their observed patients all eventually required treatment. 10 In the case of our patient, active management was chosen given the location on the face, size, and rapid growth rate. The nodular plaque appeared to respond most significantly to the combination of oral steroids and intralesional 5-FU injections.
This case emphasizes the clinical variability of RDD and underscores the importance of histopathological evaluation to help clarify diagnosis, investigations, and guide management for patients presenting with unusual clinical variants. It is not known at what point, or if there is a risk of progression from CRDD to systemic disease and longitudinal follow-up of patients with CRDD is likely beneficial to establish more effective treatments and better characterize the long-term sequalae of the condition.
Footnotes
Acknowledgements
The authors sincerely thank Dr. Stephanie Petkiewicz for her assistance, guidance, and review of the histopathological components of this case report. The authors declare that the contents of this article are their own original unpublished findings and did not receive any funding for the writing of this manuscript. All authors have contributed significantly and agree with the content of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
The patient provided informed consent for publication of the case report and images.