
Abstract
Rosai–Dorfman disease is a rare reactive histiocytic proliferation disorder. It is rarely reported in the literature, and its clinical manifestations vary depending on the location of occurrence. The clinical manifestations and imaging features are not characteristic, and the diagnosis mainly relies on pathology and immunohistochemistry. Here, we report a case involving the nasal cavity and paranasal sinuses, which did not exhibit typical symptoms such as painless cervical lymphadenopathy and fever. It could easily have been misdiagnosed as a malignant tumor before surgery. This case did however exhibit typical histopathological features. The patient was treated with surgery combined with hormone therapy and a good clinical outcome was achieved.
Keywords
Introduction
Rosai–Dorfman disease (RDD) is a rare non-genetic disease with an unclear etiology, which may be related to viral infection, genetic factors, immune deficiency, or tumors. RDD mostly occurs in adolescents. According to the lesion sites involved, it exhibits different clinical manifestations, but lacks characteristic signs and symptoms, which easily leads to misdiagnosis or missed diagnosis. The typical pathological feature of RDD is the infiltration and phagocytosis of lymphocytes observed by histopathology. Immunohistochemistry demonstrating positivity of S100 and CD68 in tissue cells is a necessary diagnostic indicator. Treatment methods include surgical resection, hormone therapy, radiotherapy, chemotherapy, immunotherapy, and targeted therapy. In this article, we report a case of RDD affecting the nasal cavity and paranasal sinus, and through the combination of this case and relevant literature review, we discuss the clinical manifestations, diagnosis, and treatment of this disease, with the aim of providing reference data for clinicians and improving the diagnosis rate of RDD.
Case report
A 52-year-old female patient, was admitted to the hospital with nasal congestion and dizziness for 3 months. Three months before, without any trigger, she had experienced the onset of bilateral nasal congestion, which was intermittent and gradually worsened, accompanied by dizziness, and reduced sense of smell, but without nasal itching, sneezing, or clear nasal discharge. There was no nasal bleeding, blood in the nasal discharge; no headache, fever, or vomiting. The patient was treated with nasal drops and oral medication, but the symptoms did not improve. Physical examination revealed a perforation of the nasal septum, and the anterior segment of the right nasal cavities was filled with a dark red neoplasm, which was relatively tough, with unclear boundaries and a rough surface, which bled easily when touched (Figure 1). A computed tomography (CT) scan of the paranasal sinuses showed the presence of patchy soft tissue shadows in the right nasal cavities, with bone destruction in the anterior part of the nasal septum adjacent to it (Figure 2). After admission, a nasal endoscopy-assisted nasal tumor resection was performed. During the operation, dark red neoplasms were found in the anterior segment of the right nasal cavities, with unclear boundaries, uneven surfaces, and relatively tough texture. The tumor tissue had caused damage to the anterior part of the nasal septum, the middle turbinate, and the uncinate process, and the landmarks were unclear. Postoperative pathological examination revealed the “invasion phenomenon” (Figure 3). Postoperative immunohistochemical analysis showed: S100 (+), CD68 (+), CD1a (−), CD163 (+), K1-67 (+) ~3%, Vim (+) (Figure 4). The postoperative pathological diagnosis – right nasal cavity tumors – combined with the results of immunohistochemical examination and hematoxylin and eosin morphology, suggested that the lesion was due to RDD. Postoperative oral hormone treatment was given. The treatment plan involves administering prednisone (15 mg/day, for 10 days). Postoperative follow-up showed that the surgical area of the patient had healed well. There was no recurrence after 6 months. Currently, the patient is still under follow-up.

Nasal endoscopy revealed a dark red neoplasm with an uneven surface filling the anterior nasal cavity (arrow).
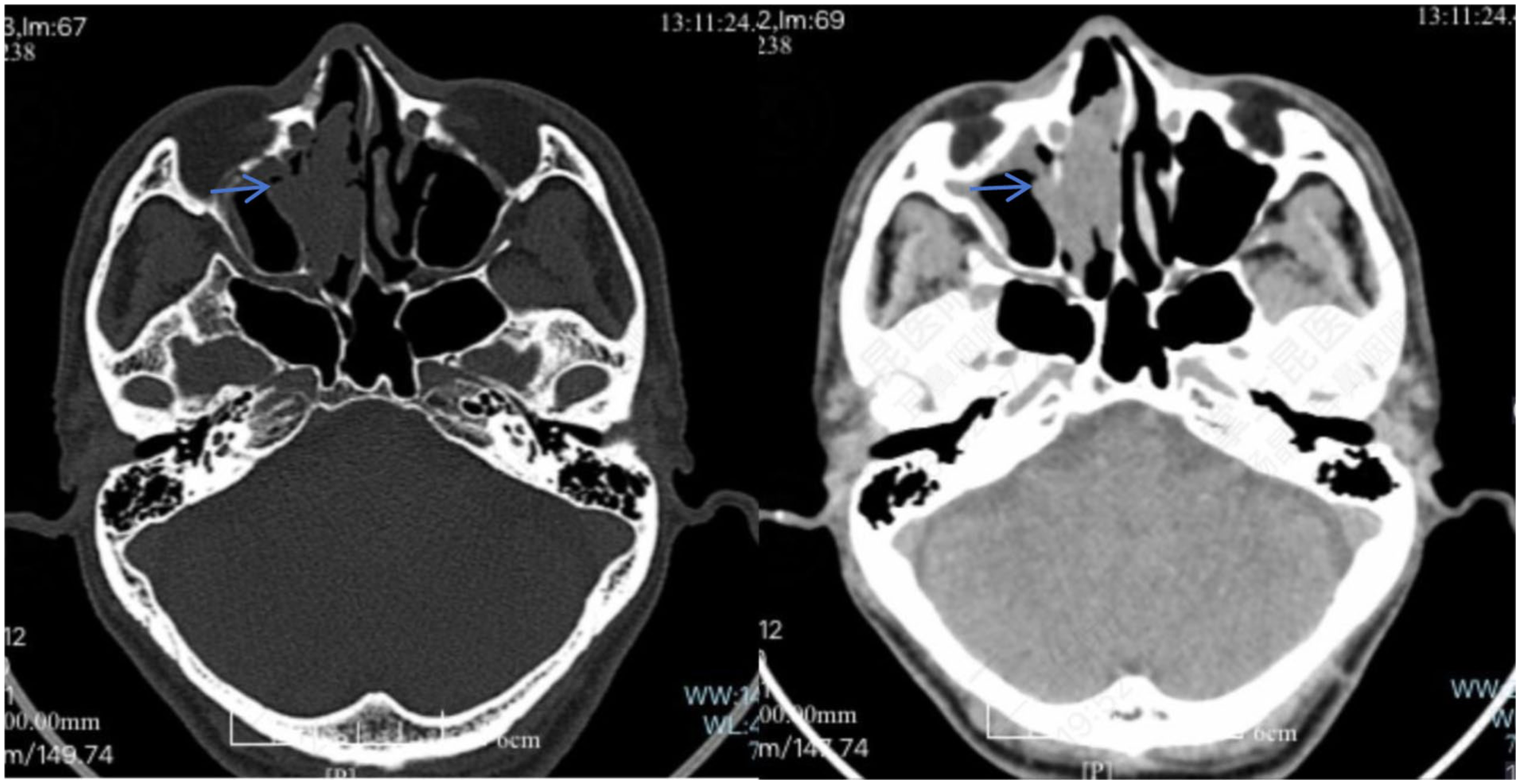
A CT scan of the paranasal sinuses showed the presence of patchy soft tissue shadows in both nasal cavities, with bone destruction in the anterior part of the nasal septum adjacent to it (arrow).
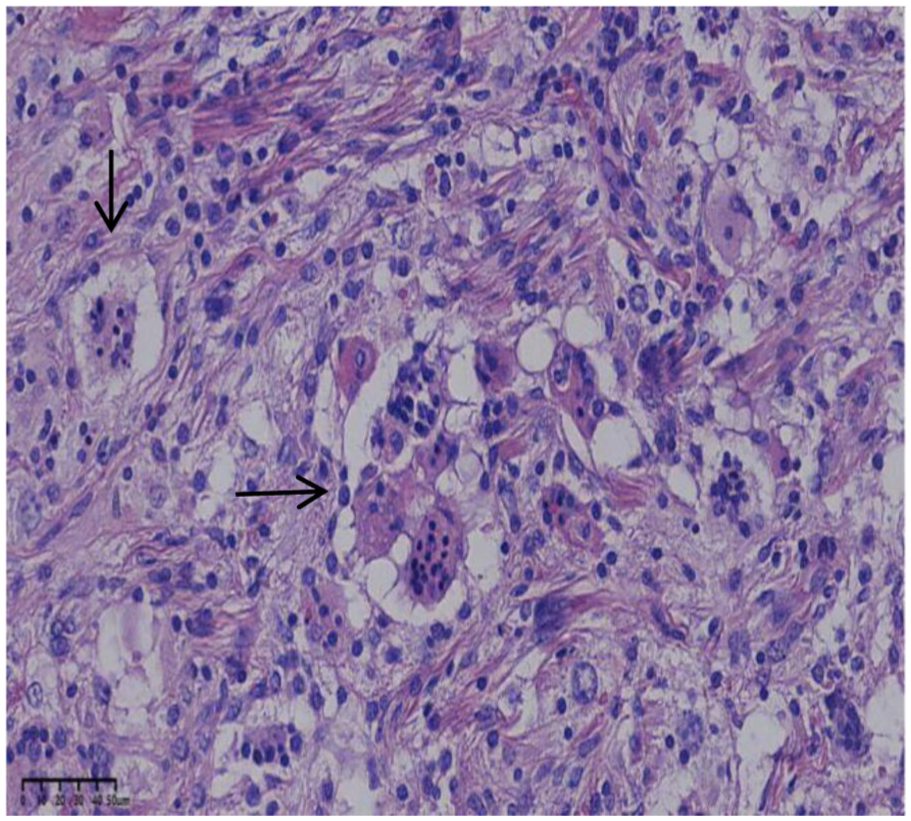
Postoperative pathological examination showed “infiltration phenomenon” (arrow).

Immunohistochemical examination: (a) CD68 positive in the cytoplasm of histiocytes (×200), (b) CD1a negative in histiocytes (×200), and (c) S-100 positive in the cytoplasm of histiocytes (×200).
Discussion
RDD is also known as sinus histiocytosis with massive lymphadenopathy. It was first described in 1969 by Rosai and Dorfman as a benign proliferative lesion of tissue cells.1,2 It was previously considered a rare benign disease of hematopoietic tissue cells, but most current studies suggest that RDD is a clonal tumor disease. 3 It is more common in adolescents, with no difference in incidence between males and females. 4 In 2016, the revised classification criteria for histiocytosis classified RDD as Group R, which can be divided into lymph node type, extranodal type, familial type, tumor-related type, and autoimmune-related type. 5 In the latest fifth edition of the World Health Organization classification of lymphoid and hematopoietic tumors and the international consensus classification, RDD was first classified under the chapter of histiocyte/macrophage tumors. 6
The pathogenesis of RDD is unclear and may be related to viral infection, genetic factors, immune deficiency, and tumor-related factors. In recent years, it has been reported that about one-third of patients have gene mutations involving the MAPK/ERK pathway, 7 such as mutations of KRAS, NRAS, HRAS, ARAF, BRA, and MAP2K1. The abnormal activation of this pathway can cause cells to lose their ability to undergo apoptosis and differentiation, causing abnormal cell proliferation and malignant transformation. 8
The clinical manifestations include bilateral painless cervical lymph node enlargement, along with the possible involvement of other lymph node sites such as the axilla, inguinal region, and mediastinal lymph nodes. Local swelling and pain may also occur, accompanied by increased erythrocyte sedimentation rate, and increased numbers of white blood cells. Some patients may also have fever, weight loss, anemia, and discomfort. The most commonly affected organs include the respiratory tract, sinuses, skin, bones, central nervous system, urogenital tract, orbit, and soft tissues. For RDD involving the nasal cavity and sinuses, the clinical manifestations include nasal congestion, dry nose, reduced sense of smell, runny mucus, head and face pain, and external nose swelling. For RDD involving the nasal septum, nasal cavity, sinuses, inferior turbinate/median turbinate, and cervical lymph nodes, however, the clinical manifestations of this patient were nasal congestion, without bilateral painless cervical lymph node enlargement. A CT scan showed a patchy soft tissue shadow in the right nasal cavity, with bone destruction in the anterior part of the nasal septum adjacent to it. RDD has no characteristic clinical manifestations or imaging features. In addition, some inflammatory or tumor-related changes in the same location may lead to misdiagnosis or missed diagnosis. 9
Currently, histopathology is the gold standard for diagnosing RDD. A clear pathological feature of RDD is lymph node enlargement accompanied by diffuse infiltration of tissue cells. Typical lesions show “invasion phenomenon” or “lymphocyte phagocytosis,” that is, the presence of lymphocytes, plasma cells, and neutrophils in varying numbers and exhibiting complete morphology within the tissue cells, which are the characteristic pathological manifestations of this disease. However, the invasion phenomenon is not a necessary condition for the diagnosis of RDD. The most important diagnostic basis is immunohistochemistry, demonstrating expression of S100, CD68, and CD163, as well as BCL-1 and OCT2, while CD1a is negative. 10
There is currently no unified treatment plan for RDD. It is recommended to adopt individualized treatment with the aim of controlling symptoms. For patients without symptoms or with mild symptoms, observation can be performed first. Currently, no international standardized treatment for RDD has been determined. The “NCCN Oncology Clinical Practice Guidelines for Tissue Cell Tumors, Second Edition 2021” proposed diagnosis and treatment procedures for RDD patients, but for single cases of extranodal or intranodal RDD, surgical resection is both a method for clear diagnosis and a treatment method. Surgical resection can cure single-focal diseases. For patients who cannot undergo surgical treatment or have recurrent/refractory cases, corticosteroids can be used to reduce the size of the lesion and improve symptoms. Radiotherapy, chemotherapy, immunotherapy, and targeted therapy can also be used to alleviate local symptoms.11,12 For this patient in this study, a surgical resection was performed. After the surgery, she was orally administered the hormone prednisone for 10 days. Currently, there has been no recurrence after 6 months, and she is still under follow-up.
In conclusion, RDD is a rare benign and self-limiting histiocytic disorder. Its clinical and imaging manifestations lack specificity, and diagnosis mainly relies on pathological examination and immunohistochemistry. The phenomenon of phagocytosis by lymphocytes or their deep movement are typical pathological features. Imaging examinations are used to determine the surgical resection range. Surgery combined with glucocorticoid treatment yields a favorable long-term outcome. Cases of this disease are relatively rare, and more cases need to be collected for systematic research and analysis to identify the best treatment plan. This case report and literature review are presented for reference and learning purposes. It is hoped that enabling clinicians to fully understand RDD will improve the diagnosis rate of the disease and provide the best treatment plan for patients.
Footnotes
Acknowledgements
The authors would like to thank Prof. Guojing Zhao from the Department of Pathology, The First Affiliated Hospital of Kunming Medical University for providing and reviewing the pathological image.
Ethical considerations
The authors’ institution does not require ethical approval for reporting individual cases or case series.
Consent for publication
Written informed consent was obtained from the patient for anonymized patient information to be published in this article.
Author contributions
Guifeng Yang: data collection. Jing Yang: writing. Both authors reviewed the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data underlying the results are available as part of the article and no additional source data are required.