
Abstract
Amphiregulin (AR), belongs to the epidermal growth factor (EGF) family, is able to induce a series of pathological and physiological responses by binding and activating epidermal growth factor receptor (EGFR). Interleukin-8 (IL-8) or CXCL8, a pro-inflammatory chemokine, has been suggested to be involved in tumor cell proliferation and inflammatory microenvironment via transactivation of the EGFR. However, whether there is a crosstalk between AR with IL-8 during inflammatory response remain to be fully understood. The current study was designed to investigate the possible mechanism of the interactions between AR and IL-8 production in human lung epithelial cells in vitro. Lung epithelial A549 cells were stimulated with lipopolysaccharide (LPS) to generate ALI model. LPS-induced AR and IL-8 production by A549 cells was measured by real-time PCR, Western Blot, and ELISA. The AR neutralizing antibody, PI3K specific inhibitor LY294002, JNK specific inhibitor SP60012, ERK specific inhibitor PD98089, and p38 inhibitor SB203580 were used to investigate the role of these signal pathways in LPS-induced cell proliferation, AR and IL-8 expression. LPS could induce AR through PI3K/Akt and ERK signal pathways. Furthermore, LPS induced AR promoted the production of IL-8 requires activation of EGFR, PI3K/Akt, and ERK signal pathways. The neutralizing antibody to AR prevented production of IL-8 induced by LPS. Treatment with Erlotinib, PI3K inhibitors, ERK inhibitor significantly inhibited AR-induced IL-8 production and cell proliferation. Our data indicate that a distinct role of EGFR–PI3K–Akt/ERK pathway as a bridge of interaction between AR and IL-8 production, as one of potential mechanisms to regulate inflammation and cell proliferation in human lung epithelial cells.
Introduction
EGFR as a growth-factor-receptor tyrosine kinase glycoprotein, a member of the HER/ErbB protein family, consisting of an extracellular ligand-binding domain, tyrosine kinase activity, capability of autophosphorylation, and a transmembrane tail with an ATP-binding site. 1 A growing amount of evidence indicates that EGFR is closely related to lung development 2 and a rising star in the era of precision medicine of lung cancer 3 by affecting a multiplicity of signal transduction pathways like cellular proliferation, migration, neoangiogenesis, adhesion, and inflammatory response.4–6 Numerous ligands can bind with EGFR, such as amphiregulin (AR), epiregulin (ER), transforming growth factor-α (TGF-α), EGF, heparin-binding EGF-like growth factor (HB-EGF), betacellulin (BTC), and neuregulin (NRG) subfamily. 7 When EGFR is activated, the intrinsic kinase activity leads to homo- or hetero-dimerization of the receptor with another member of the HER/ErbB family, then EGFR tyrosyl-phosphorylation and allow proteins to bind through their Src homology 2 (SH2) domains leading to the activation of downstream signaling cascades such as the Ras/mitogen activated protein kinase (MAPK) pathway and the phosphoinositide-3 kinase (PI3K)/Akt pathway.8–10
AR has a higher affinity and specificity for EGFR,11,12 which can activate different signal transduction pathways, including RAS/RAF/MEK/ERK and PI3K/Akt, and induce a series of biological effects. 13 AR was reported to be up regulated in those damaged lung tissues of the patients with different pulmonary diseases, such as asthma, chronic obstructive pulmonary disease and in lung epithelial cells in a ventilator-associated lung injury model. 14 It has been reported that AR-EGFR pathways are involved in orchestrating immunity, inflammation, and tissue repair. 15 AR-producing pathogenic memory Th2 cells can control airway fibrosis by inducing inflammatory eosinophils to secrete osteopontin. 16 Additionally, AR binds to the EGFR and activates downstream proteins, such as MMP-13, 17 COX-2, 18 and GM-CSF, 19 to induce a series of pathological processes, such as inflammatory diseases, rheumatoid arthritis, and development of tumors .
The previous study on particulate matter with an aerodynamic diameter <2.5 µm (PM2.5) with inflammation of airways demonstrated that AR played an important role in sustaining proinflammatory response and was related to the disease severities. 19 AR can induce IL-8, COX-2, and VEGF secretion in airway epithelial cells. 20 They also found that cigarette smoke-induced IL-8 expression by lung epithelial cells involves transactivation of the EGFR and that autocrine production of EGFR ligands make a substantial contribution to this response. 21 Additionally, emerging data suggested that EGFR–PI3K-Akt/ERK pathway was involved in the development of lung cancer inflammatory microenvironment by the hyper-production of IL-8.22,23 All these studies suggested that AR and IL-8 are associated with inflammatory microenvironment. However, the crosstalk between AR and IL-8 during lung inflammation remains to be fully understood.
The present study is aim to explore the potential association and interaction mechanisms between AR and IL-8 in the inflammatory response, further to investigate the expression and biological function of AR protein and its receptor in lung epithelial cells, and to define the role of AR in the regulation of IL-8 production in lung epithelial cells. Furthermore, this study investigated the involvement of EGFR–PI3K-Akt/ERK pathway activation in AR induced IL-8 production and lung epithelial cells proliferation.
Materials and methods
Cell lines and reagents
Human lung cancer cell line A549 cells were purchased from Shanghai Institute for Biological Science and cultured at 37°C in a 5% CO2 atmosphere in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin(100 U/ml), and streptomycin (100 mg/ml). Human recombinant AR, ELISA kits for IL-8, anti-human AR neutralizing antibody were purchased from R&D Systems (Shanghai, China). Specific PI3K inhibitor LY294002, specific ERK inhibitor PD98059, specific p38 kinase inhibitor SB203580 and specific JNK inhibitor SP600125 were purchased from Biovision Company (California, USA). EGFR inhibitor Erlotinib was purchased from Roche Company (Basel, Switzerland). EGFR antibody for immunofluorescent staining was purchased from Abcam (HK, China).
Quantitative RT-PCR
Total RNA was extracted from A549 cells using TRIzol reagent (Invitrogen, USA) and reverse transcribed to cDNA with the SuperScript First-strand Synthesis System (Invitrogen, USA). Quantitative RT-PCR was carried out using an ABI Prism 7000 PCR instrument (Eppendorf, Germany) with SYBR Green PCR master kit according to the manufacturer’s instructions. The primers used were as follows: AR: 5′-CAGTGCTGATGGATTTGAGGT-3′ and 5′-TAGCCAGGTATTTGTGGTTCG -3′; ER:5′-TCCATCTTCTACAGGCAGTCC-3′ and 5′-TATTGACACTTGAGCCA CACG-3′; EGF:5′-CTCATCACTGGTTGTGGTTCAT-3 and 5′-CATAAAACCTTC ACGACACGAA-3′; TGF-α: 5′-CCTGGCTGTCCTTATCATCAC-3′ and 5′-ACT GTTTCTGAGTGGCAGCA-3′; HB-EGF: 5′-C CTATGACCACACAACCATCC-3′ and 5′-TGCCCAACTTCACTTTCTCTT-3′; BTC: 5′-TGAAACTAATGGCCTC CTCTGT-3′ and 5′-CTTTTACGACGTTTCCGAAGAG-3′; NRG: 5′-GGATTCAC TGGAGCAAGATGT-3′ and 5′-CGGTTATGGTCAGCACTCTCT-3′; and GAPDH: 5′-CCACCCATGGCAAATTCCATGCA-3′ and 5′-TCTACACGGCAGGTCAGGT CCACC-3′. For data analysis, the raw cycle threshold (CT) value was first normalized to the GAPDH gene for each sample to get △CT. The normalized △CT was then calibrated to control cell samples to get △△CT.
Western blot analysis
To measure the expression of AR and the signal pathway induced by LPS, A549 cells were cultured in six well plate (1 × 105 cells/well) for 24 h and treated with LY294002, SP600125, SB203580, and PD98059 at 10, 20, and 30 µM for another 2 h. Then cells were stimulated with or without LPS (Escherichia coli, 055:B5, Sigma) at 1 µg/ml for 24 h. Then intracellular protein was extracted by RIPA lysis buffer and western bolt analysis was performed as described previously. All results were calculated by Phoretix 1D software.
Enzyme-linked immunosorbent assay (ELISA)
A549 cells were cultured in 24 well cell culture microplates (1 × 105 cells/well) for 24 h and then treated with LPS at concentrations of 0.01, 0.1, and 1 µg/ml for another 24 h. Levels of IL-8 in supernatant were measured by ELISA, according to the manufacturer’s instructions. Next, cells were pre-incubated with an anti-human AR neutralizing antibody at a concentration of 1, 10, 20 µg/ml, 2 h before LPS stimulation. After that, A549 cells were treated with AR at 40 ng/ml alone or in combination with five inhibitors (Erlotinib, LY294002, SP600125, SB203580, and PD98059) for 24 h. IL-8 in supernatant was measured by ELISA as mentioned above. Each experiment was done in six replicate wells for each drug concentration and the standard deviations were calculated accordingly.
Immunofluorescence staining
A549 cells were seeded onto sterile coverslips placed in 24-well cell culture plates (at a concentration of 1 × 104 cells/ml/well) and allowed to grow for 24 h. Then, cells were fixed with 4% paraformaldehyde for 20 min at room temperature, washed three times in PBS and permeabilized by 0.1% Triton X-100 for 20 min at room temperature. Subsequently, the cells were blocked for 30 min in PBS containing 10% goat serum after three washes in PBS. Next, the cells were incubated overnight with mouse monoclonal anti-His tag antibody diluted in PBS/10% goat serum (1:40) (Abcam, HK, China), and FITC conjugated anti-mouse IgG antibody (Abcam, HK, China) diluted in PBS/10% goat serum (1:100) for 1 h at room temperature. After three washes in PBS and counterstaining with DAPI (4, 6′-diamidino-2-phenylindole) for 5 min, cells were examined for under immunofluorescence microscope (Olympus, Japan).
Cell proliferation assay
Cells (1 × 104 cells/well) were seeded in 96-well plates, treated with AR (10, 20, 30, or 40 ng/ml) for 24, 48, or 72 h. Cells (1 × 104 cells/well) were seeded in 96-well plates in the absence or presence of Erlotinib, LY294002, or PD98059 and incubated with AR (40 ng/ml) for 24, 48, or 72 h. Then, 10 μl of the Cell Counting Kit-8 solution (Dojindo, Japan) was added to the medium. After incubating for 2 h, the amount of orange formazan dye in medium was determined by measuring the absorbance at 450 nm using a microplate reader (Thermo Scientific, USA).
Statistical analysis
All data are expressed as mean ± SEM. Data were analyses by GraphPad Prism 5 software. Statistical significance was compared between groups by the Student’s t-test or ANOVA analyses. p < 0.05 was taken as statistically significant.
Results
LPS promotes AR production by human lung epithelial cells
To address whether there are multiple EGF-related growth factors that contribute to EGFR-mediated cell proliferation and IL-8 production. We examined the mRNA expression of the seven EGFR ligands in LPS-treated A549 cells. Among the seven EGFR ligands we examined, AR expression were significantly upregulated by LPS stimulation at concentration of 1 µg/ml (p < 0.01, Figure 1(a)). AR mRNA expression in LPS-stimulated A549 cells rise significantly at 4 h and continues to peak at 24 h (p < 0.05 or 0.01, respectively, Figure 1(b)). Furthermore, AR protein expression were upregulated at 12 h and continued to increase in intensity up to 48 h (Figure 1(c) and (d)). These results suggest that AR gene and protein show a gradual and sustained activation in A549 cell after LPS stimulation.

LPS induces increased production of AR in human lung epithelial cells: (a) mRNA expression of EGFR ligands from A549 in response to LPS at 4 h, (b) A549 cells were stimulated with LPS (1 µg/ml) for indicated times. Real time–PCR shows LPS-induced expression of AR mRNA with a peak at 4 h of stimulation and continues to peak at 24 h, and (c and d) LPS induced AR protein expression for indicated times. Each data point represents mean ± SEM of three experiments.
PI3K and ERK are involved in LPS-induced AR expression in human lung epithelial cells
The stimulation of TLR4 by LPS induces the activation of PI3K and MAP kinases including JNK, ERK, and p38 kinase. The PI3K specific inhibitor LY294002, JNK specific inhibitor SP60012, ERK specific inhibitor PD98089 and p38 inhibitor SB203580 were used to investigate the role of these signal pathways in LPS-induced AR expression. As shown in Figure 2. Interestingly, LPS-induced AR expression at 24 h after LPS stimulation was significantly decreased by PI3K specific inhibitor LY294002 (Figure 2(a)) and ERK specific inhibitor PD98089 (Figure 2(d)) treatment (p < 0.05 or 0.01, respectively), but not by p38 inhibitor SB203580 (Figure 2(b)) and JNK specific inhibitor SP60012 (Figure 2(c)) treatment. These results demonstrate that LPS-induced PI3K and ERK activation is associated with significantly increased AR expression in human lung epithelial cells.

Involvement of PI3K and MAPK pathways in the regulation of AR expression. A549 cells were pretreated with DMSO, PI3K specific inhibitor LY294002 (LY, a), p38 inhibitor SB203580 (SB, b), JNK specific inhibitor SP60012 (SP, c) and ERK specific inhibitor PD98089 (PD, d) at various concentrations for 2 h and then stimulated with 1 µg/ml LPS for 24 h. Expression level of AR was examined by Western blot. Each data point represents mean ± SEM of three experiments.
AR induced by LPS further stimulates IL-8 production in human lung epithelial cells
As mentioned above, LPS can induce AR expression in lung epithelial cells. Similarly, IL-8 secretion was also increased significantly in A549 cells stimulated by LPS. 24 However, whether there is a crosstalk between AR and IL-8 which might regulate inflammatory microenvironment remain to be fully understood. To address this question, A549 cells were pretreated with AR neutralizing antibody, then IL-8 production was measured by Real-time PCR (Figure 3(a)) and Western blot (Figure 3(b)). Results suggested that AR neutralizing antibodies at the concentration of 10 or 20 µg/ml blocked LPS-induced IL-8 production (p < 0.05 or 0.01, respectively). To characterize the individual contribution of AR on IL-8 production, we further stimulated the A549 cells with human recombinant AR. Consistent with the above data, AR at the concentration of 40 ng/ml could significantly increase both IL-8 mRNA (Figure 3(c)) and protein (Figure 3(d)) expression in lung epithelial cells. Our results demonstrated that AR may directly and efficiently stimulate IL-8 production of lung epithelial cells in a dose depended manner.

LPS-induced IL-8 production is AR dependent in A549. A549 cells were pretreated with AR neutralizing antibody for 2 h and then stimulated with LPS (1 µg/ml) for 4 h: Total RNA was extracted and subjected to reverse transcription followed by qPCR to detect IL-8 mRNA expression (a), IL-8 in cell-free supernatants after 24 h stimulated by LPS were assayed ELISA (b), A549 cells were then stimulated with AR (10, 20, and 40 ng/ml) for 4 h to detect IL-8 mRNA (c), or 24 h to detect its secretion of IL-8 in supernatants (d). Each data point represents mean ± SEM of three experiments.
Expression of EGFR in human lung epithelial cells
AR displays higher affinity and specificity for EGFR. Whether EGFR is expressed on human lung epithelial A549 cells remain unclear. Immunofluorescence staining were used to analyze EGFR expression in A549 cells. As show in Figure 4(a), A549 cells can constitutively express EGFR, which indicates that lung epithelial cells may be responsive to AR. Then, we stimulated A549 cell with different concentrations of AR, the EGFR and p-EGFR were measured by western blot. As show in Figure 4(b) and (c), AR could induce EGFR Phosphorylation (p-EGFR) on A549 cells at the concentration of 20 ng/ml or 40 ng/ml.

Expression and activation of EGFR on A549: (a) A549 cells were positive for EGFR (red labeling) after stimulation with AR (40 ng/ml) for 24 h in immunofluorescence analysis. DAPI staining (blue) indicates the localization of nuclei. One representative image from three independent experiments is presented and (b and c) EGFR phosphorylation were measured by western blot. AR could induce EGFR Phosphorylation (p-EGFR) on A549 cells at the concentration of 20 ng/ml or 40 ng/ml. Each data point represents mean ± SEM of three experiments.
Effects of EGFR, PI3K, JNK, P38, and ERK inhibitors on AR stimulated IL-8 production
As show in Figure 3(b) and (d), AR stimulated a significant increase of IL-8 production from A549 cells in a concentration-associated pattern, as compared to cells without AR stimulation at 24 h (p < 0.05 or 0.01, respectively). From our pilot study, we found AR at 40 ng/ml increased IL-8 production from 12 h, which could maintain during 48 h (data not shown). Treatment with Erlotinib (TKI) at all concentrations significantly reduced AR-stimulated IL-8 production, as compared to cells treated with AR alone (p < 0.05 or 0.01, respectively, Figure 5(a)). Treatments with LY294002 at 20 and 30 µM or PD98059 at 30 µM significantly inhibited AR-induced IL-8 production (p < 0.05 or 0.01, respectively, Figure 5(b) and (e)). Levels of IL-8 in the supernatant of cells with LY294002 at 10 µM, PD98059 at 10 and 20 µM, or SP60012 (Figure 5(c)) and SB203580 (Figure 5(d)) at all concentrations were still significantly higher than that of cells treated with DMSO only (p < 0.05 or 0.01, respectively).
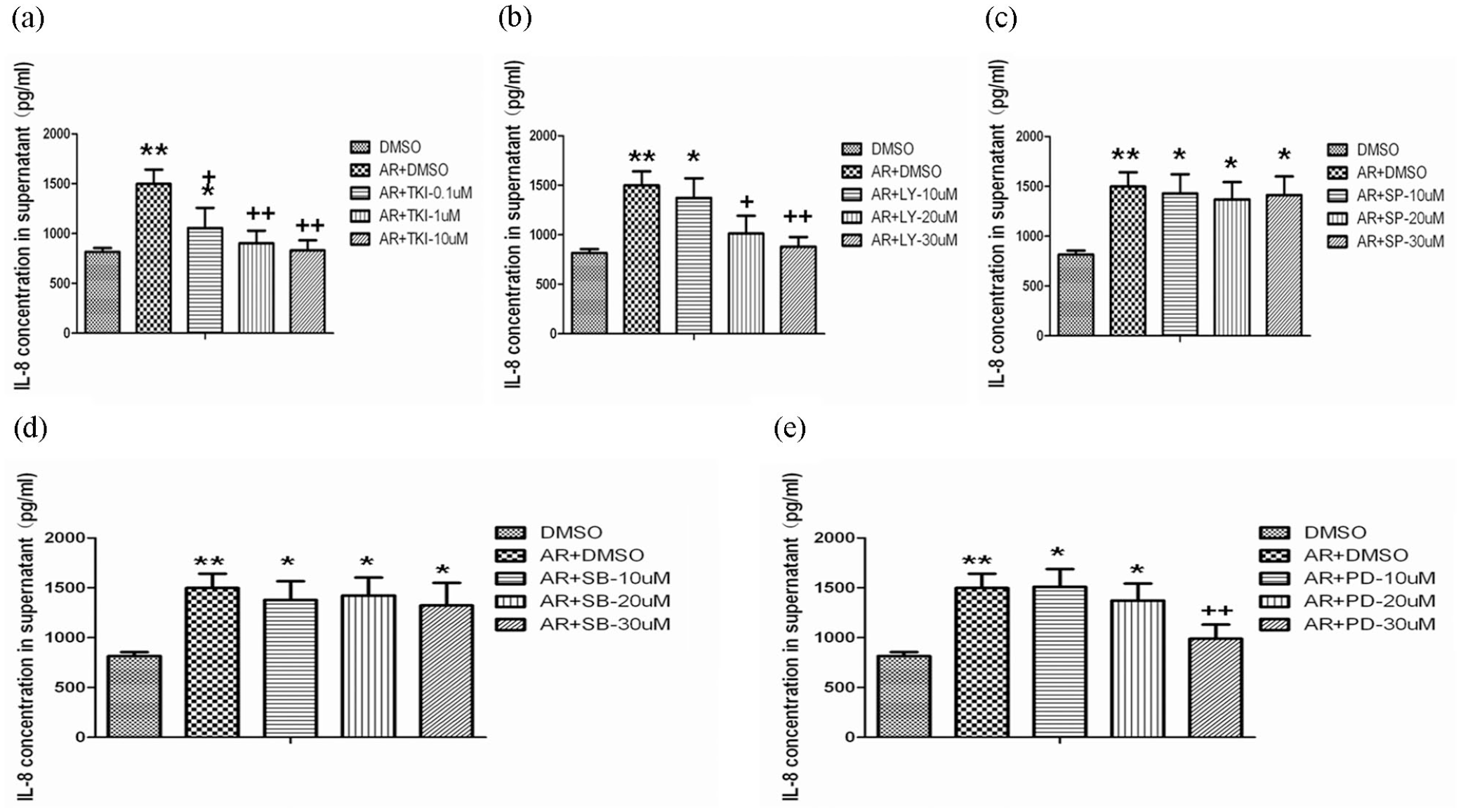
Effects of EGFR, PI3K, JNK, P38, and ERK inhibitors on AR stimulated IL-8 production: IL-8 production from cells was measured 24 h after the culture with DMSO alone, AR at 40 ng/ml plus DMSO, Erlotinib (TKI; a) at doses of 0.1, 1.0, or 10 µM, or LY294002 (b), JNK specific inhibitor SP60012 (c), p38 specific inhibitor SB203580 (d), and ERK specific inhibitor PD98089 (e) at doses of 10, 20, or 30 µM. Data is presented as mean ± SEM and each group have six measurements.
Inhibitory effects of EGFR, PI3K, and ERK inhibitors on AR stimulated cell proliferation
A549 cells were stimulated with AR (10, 20, and 40 ng/ml) for different time point. The percentage of total cell number significantly increased after the stimulation of AR at 20 or 40 ng/ml for 72 h, as compared to DMSO alone (p < 0.05 or 0.01, respectively, Figure 6(a)), which was significantly inhibited by TKI (Figure 6(b)), LY294002 (Figure 6(c)) or PD98059 (Figure 6(d)) at various concentrations. The inhibitory effects of inhibitors (LY294002 and PD98059) showed a dose-dependent pattern.

The increased percentage of the total number of A549 cells was assessed using Cell Counting Kit-8 solution: the percentage of total number of cells significantly increased after the stimulation with AR (40 ng/ml), compared to the average of total cells values treated with vehicle (DMSO) alone (a), while cells treated with Erlotinib (b) at doses of 0.1, 1.0, or 10 µM, or LY294002 (c), and PD98059 (d) at doses of 10, 20, 30 µM, showed a lower increase in the percentage of total number of cells, respectively. Data were presented as mean ± SEM and each group have six measurements.
Discussion
AR was found to be expressed in the alveolar and airway epithelium, macrophages, and fibroblasts.25,26 The evidence from previous studies suggested that AR plays a critical role in the development of lung inflammation through the over-production of IL-8 by binding to EGFR.27,28 It has been reported that the activation of the EGFR pathway also could induce IL-8 secretion in human bronchial epithelial cells by multi-stimuli, such as EGF, 22 BTC, 4 HB-EGF, 29 and MMP-12. 30 Results from this study demonstrated that AR may directly and efficiently stimulate IL-8 production of lung epithelial cells in a dose-dependent manner, which was inhibited by EGFR inhibitor. The present study also demonstrated that the epithelial cells may act as a primary receptor to be stimulated by LPS, and as the secondary reactor to regulate the over-production of IL-8 through EGFR-PI3K-Akt/ERK pathway, and further to accelerate the local inflammatory response. It’s indicated that the signal pathway of AR-EGFR-PI3K-Akt/ERK plays the crucial role in the mechanism of IL-8 production in lung epithelial cells, which helps to understand the potential of new anti-inflammatory therapeutic target in acute lung injury or other pulmonary inflammatory diseases.
LPS can produce different inflammatory mediators by incubation with lung epithelial cells. Alveolar type II cell injury is a key factor in the pathogenesis of ALI. 31 Therefore, A549 cells, which are well-established EMT and acute lung injury cell model in many articles, 32 were used in the in vitro experiments in the present study. There are still some differences between tumor cells and normal alveolar epithelial cells. In the future, we can try to perform relevant experiments on primary alveolar epithelial cells and animal model.
In this study, we examined several EGFR ligands in lung epithelial cells A549, and found that the AR, ER, and BTC could be upregulated by LPS, but only AR show a strong stimulus response, which could be blocked by PI3K and ERK inhibitor. Our study also indicated that both endogenous and exogenous AR could promote the IL-8 secretion. We found that the levels of IL-8 partially blocked by AR neutralizing antibody, and its expression in lung epithelial cells challenged with LPS were still significantly higher than those without LPS, which suggest that there are some other EGF-related growth factors, like BTC 4 and EGF, 22 may participate in such biological efforts. Our study demonstrated that the AR-EGFR-PI3K-Akt/ERK signal pathway may play the decisive role in the mechanism of IL-8 production and could be the potential of new anti-inflammatory therapeutic target in lung epithelial cells, evidenced by the finding that the over-production of IL-8 induced by AR was fully prevented by EGFR, PI3K, and ERK inhibitors.
Many EGFR-dominated signal pathways may contribute to IL-8 expression in airway epithelium cells, including PI3K, ERK, and STAT signal pathways.33,34 We provided direct evidence that lung epithelial cells A549 constitutively expressed EGFR, and the levels of phosphorylated EGFR increased after AR stimulation in human lung epithelial cells. EGFR, PI3K, and ERK inhibitor could inhibit over-production of IL-8 initiated by the over activation of AR-EGFR pathway. Continuous PI3K activation is regarded as promoting inflammation and closely associated with airway inflammatory diseases such as acute lung injury, chronic obstructive lung disease, and asthma. 35 In addition, PI3K/Akt and ERK pathways were also crucial in lung cancer cell proliferation and development. 36 Our study demonstrated that AR could increase the proliferation of lung epithelial cells, which could be down regulated by EGFR, PI3K, and ERK inhibitors. We initially found that LPS stimulated lung epithelial expression of AR is functionally active, which could directly stimulate IL-8 production and cell proliferation through the activation of EGFR, PI3K/Akt, and ERK signal pathway, and then lead to the recruitment of inflammatory cells in the lung tissue and the formation of inflammatory microenvironment.
Our study demonstrated that EGFR inhibitor could inhibit IL-8 production and cell bio-behaviors even at the lowest dose (0.1 µM) and did not show the dose-associated pattern in the range of concentrations used in the present study. It may be the concentration selected still high, which also indicates that this is an EGFR-dependent signal pathway model. The percentage of total cell number significantly increased after the stimulation of AR, which was partially inhibited by PI3K inhibitors (LY294002) and ERK inhibitor (PD98059) at various concentrations. It is possible that the concentration selected still low and there may be a need of even higher doses than the highest dose used in this study, or there may be multiple intracellular signal pathways involved in the signaling under the EGFR to regulate these series of biological effects.
Conclusion
Our study demonstrated that LPS can induce the over-production of AR and IL-8 from human lung epithelial cells. Both endogenous and exogenous AR could increase the over-production of IL-8, which could be blocked by EGFR, PI3K, and ERK inhibitor. The lung epithelial cells proliferation is also regulated by EGFR-PI3K-Akt/ERK pathway activation. Thus, our data indicate that the AR-EGFR–PI3K-Akt/ERK loop-dependent pathways play an important role in the inflammatory response and cell proliferation, as a novel therapeutic approach to lung inflammatory diseases.
Supplemental Material
sj-tif-1-eji-10.1177_2058739221998202 – Supplemental material for Amphiregulin induces interleukin-8 production and cell proliferation in lung epithelial cells through PI3K-Akt/ ERK pathways
Supplemental material, sj-tif-1-eji-10.1177_2058739221998202 for Amphiregulin induces interleukin-8 production and cell proliferation in lung epithelial cells through PI3K-Akt/ ERK pathways by Fangfang Yang, Wei Xu and Yanli Pei in European Journal of Inflammation
Footnotes
Author contributions
Conceived and designed the experiments: F.Y., Y.P., and W.X.; Performed the experiments: F.Y.; Analyzed the data: F.Y., Y.P., and W.X.; Wrote the paper: F.Y. and W.X.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by Sanming Project of Medicine in Shenzhen “the Integrated Airways Disease team led by Professor Kian Fan Chung from Imperial College London” (SZSM201612096).
Ethical approval
Ethical approval for this study was obtained from Ethics Committee of The University of HongKong-Shenzhen Hospital (APPROVAL NUMBER/ID:伦[2020]74)*.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.