
Abstract
Piperlongumine (PL), a natural alkaloid present in the fruit of the Long pepper, is known to exhibit notable anti-cancer effects. Nonetheless, the anti-tumor effect of PL in lung cancer cells still remains unclear. In the present study, we reported the chemotherapeutic effects of PL using in vitro and in vivo models. We showed that PL displayed potent anti-neoplastic activity against lung cancer A549 cells as well as corresponding docetaxel-resistant A549/DTX cells. In addition, we found that PL induced apoptosis in both A549 and A549/DTX cells. PL also induced autophagy in A549/DTX cells. Moreover, autophagy-specific inhibitors (3-methyladenine) or Beclin1 and Atg 5 small interfering RNAs (siRNAs) enhanced PL-induced apoptosis, indicating that PL-mediated autophagy may protect A549/DTX cells from undergoing apoptotic cell death. Furthermore, we observed the inhibition of phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway by PL. Finally, PL inhibited the growth of A549/DTX xenograft tumors, which was associated with inhibition of cell proliferation, induction of apoptosis of tumor cells and decreased expression of p-Akt and p-mTOR in tumor xenograft tissues. In summary, our study demonstrated that PL induced apoptosis and autophagy through modulation of the PI3K/Akt/mTOR pathway in human lung cancer cells. This study may provide a rationale for future clinical application using PL as a chemotherapeutic agent for lung cancer.
Introduction
Lung cancer is the leading cause of cancer-related mortality among malignancies worldwide. 1 In spite of significant achievement in the treatment of lung cancer over the last decade, the prognosis for patients with advanced disease remains poor. 2 Docetaxel, a semi-synthetic analog of paclitaxel, was granted approval as a first-line chemotherapy regimen for NSCLC. 3 However, chemoresistance remains a major obstacle constraining the clinical application of this agent. Therefore, it is urgent to develop novel anti-tumor agents with least side effects to meet the unmet therapeutic demand of lung cancer patients.
It is now well established that chemotherapy-induced reduction in tumor load is a function of apoptotic cell death (type I programmed cell death) involving several well-characterized morphological changes, including cell volume loss, chromatic condensation, and nuclear fragmentation. However, other research has revealed how apoptosis is attenuated in certain tumors that succeed in progressing to states of high-grade malignancy and resistance to therapy. 4 Therefore, there has been a surge of activity around identification of novel pathways of cell death, which could function in tandem in the presence or absence of efficient apoptotic machinery. In this regard, recent evidence has highlighted the existence of autophagy, or type II programmed cell death, which is activated in response to growth factor deprivation or upon exposure to anti-cancer agents. 5 Autophagy is a dynamic process involving the sequestration of cytoplasmatic portions and intracellular organelles into vacuoles called autophagosomes. These vesicles are fused with lysosomes to generate autophagolysosomes and mature lysosomes, where the sequestered material is degraded, leading to cell death. More importantly, persistent autophagy in response to cellular stress states serves as a potent death signal, as in the case of chemotherapy-induced autophagy, a specific non-apoptotic death pathway has been triggered off. However, the role of autophagy in cancer is still controversial. Recent studies suggest that autophagy is required for cancer survival 6 and tumorigenesis. 7 Autophagy contributes to cytoprotective events that help cancer cells survive under conditions of low nutrient supply and resistance to anti-cancer treatments and tumor suppression, not only by recycling of metabolites but also possibly by removing damaged organelles and growth factors and reducing chromosome instability.8,9 Autophagy may be upregulated under conditions of distress, eventually leading to cellular demise. Contribution of autophagy to programmed cell death seems to be context-dependent in various experimental systems.10–12 It is likely that apoptosis and autophagy, the two different modes of cell death, are coordinated and regulated. The cross-talk between the two pathways could be supported by the fact that various proteins encoded by the autophagy related genes (Atg) can be cleaved by apoptosis activated proteases, namely caspases and calpains. 13 The intricate relationship between apoptosis and autophagy poses a big challenge for cancer treatment.
Naturally-occurring substances are the most dependable resource for therapeutic development. A variety of FDA approved anti-neoplastic agents, including docetaxel and topotecan, have demonstrated clinical utility based on ongoing investigations of natural products. Piperlongumine (PL), a natural alkaloid of the Long pepper (Piper longum), exhibits numerous key biological activities. In addition to its insecticidal and bactericidal capabilities, 14 recent literature points to the fact that PL is capable of hindering the growth of sarcoma, bladder, breast, melanoma, and lung tumours in vitro and in vivo.15,16 The ability of PL to target the cellular stress response through direct inhibition of Glutathione S-transferase pi 1 (GSTP1) subsequently inducing of intracellular reactive oxygen species 17 accumulation promoting selective killing of cancer cells was initially demonstrated by Raj et al. 16 In addition, PL has minimal high-dose acute toxicity, and does not appear to significantly affect any biochemical, hematologic, and histopathologic parameters in animal models. Moreover, PL downregulates NF-kB activation 17 and induces rapid depletion of the androgen receptor in prostate cancer cells. 18 Furthermore, it has been reported that PL promotes autophagy in several cancer cell lines, including human PC-3 prostate cancer cells, breast cancer MCF-7 cells, renal carcinoma 786-O cells, and human osteosarcoma U2OS cells.19,20 However, the role of PL in lung cancer in this regard remains to be defined. Therefore, in the current study, we reported the anti-tumor effects and the underlying biological mechanisms of PL in lung cancer cells.
Materials and methods
Antibodies and reagents
PL was obtained from Indofine Chemical Company (Hillsborough, NJ, USA). Annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit was supplied by Biouniquer Tech (Nanjing, PR China). Cyto-ID® Autophagy detection kit was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Antibodies specific for Bcl-2, Bax, PARP, PI3K, Akt, pho-Akt (Thr308 and Ser 473), mTOR, pho-mTOR (Ser 2448), and β-actin were purchased from Cell Signaling Technology (Danvers, MS, USA). Ki-67 and the secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The protein assay kit was from Bio-Rad (Hercules, CA, USA). Akt small interfering RNA (siRNA) I kit was supplied by Cell Signaling (Beverly, MA, USA). Atg5, beclin 1 and mTOR siRNA kits were purchased from Genepharma (Shanghai, PR China). Protein inhibitor cocktail tablets were purchased from Roche Applied Science (Mannheim, Germany). Other chemicals were obtained from Sigma (St Louis, MO, USA).
Cell culture
The human lung cancer cell lines A549 was purchased from American Type Culture Collection (Rockville, MD, USA), and cultured as recommended as monolayers in DMEM medium (GibcoBRL Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; GibcoBRL Life Technologies) and 1% penicillin-streptomycin-neomycin (GibcoBRL Life Technologies), in a humidified incubator at 37°C in a 5% CO2 atmosphere. Docetaxel-resistant A549 cell line (A549/DTX) were established and preserved in 50 μg/L final concentration of docetaxel in our laboratory. Stock solution of PL (50 mM) was prepared in DMSO and all test concentrations were prepared by diluting stock solution in tissue culture medium. Cells treated with DMSO only served as a vehicle control.
MTT assay
Cell viability was analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded in 96-well plates at a density of 1000–1500 cells/well and incubated for 24, 48, or 72 h. Approximately 20 μL of MTT (5 mg/mL; Sigma, St Louis, MO, USA) was added to each well and incubated for 4 h. At the end of incubation, supernatants were removed, and 150 μL of dimethylsulfoxide (Sigma) was added to each well. The absorbance value (optical density) of each well was measured at 490 nm. All experiments were performed thrice.
Colony formation assay
Cells were plated in 6-well culture plates at 600 cells/well. Each cell group had two wells. Cells were treated with PL at the indicated dose for 48 h. After incubation for another 10 days at 37°C, cells were washed twice with phosphate buffered saline and stained with hematoxylin solution. The number of colonies containing >50 cells was counted under a microscope. The colony formation efficiency was calculated as (number of colonies/number of cells inoculated) × 100%. All assays were independently performed in triplicate.
Detection of apoptosis
PL-induced apoptotic cell death in A549 and A549/DTX cells was quantitatively determined by flow cytometry. Briefly, after treatment of cells with different concentrations of PL (6 and 10 µM) for 48 h, cells were harvested, washed with PBS, and stained with FITC Annexin V Apoptosis Detection Kit (BD Biosciences), and analyzed on an Accuri C6 Flow Cytometer.
Cyto-ID staining assay
Cyto-ID is a proprietary reagent, which specifically labels autophagic vacuoles and co-localizes with light chain 3 (LC3). Cyto-ID® Autophagy detection kit (Enzo Life Sciences) was used according to the manufacturer’s protocol. The fluorescence was measured by Tecan GeNios microplate reader (Tecan, Finland).
Monodansylcadaverine staining assay
Monodansylcadaverine (MDC), a lysosomotropic fluorescent compound, which selectively labels autophagic vacuoles, was used to assess autophagy induction as previously reported. 21
Western blot analysis
A549 and A549/DTX cells were treated with PL (0, 6, and 10 μM) for 48 h, cells were harvested, washed with cold PBS, and lysed with ice-cold lysis buffer supplemented with protease inhibitors. Equivalent amounts of protein (30 μg) from each cell lysate were resolved on SDS-PAGE. Gels were electroblotted onto nitrocellulose membranes (0.45 μM; Bio-Rad), which were then incubated in blocking solution (1×PBS, 0.1% Tween-20, 5% non-fat dry milk powder) for 1 h at room temperature. Membranes were incubated with the primary antibody at 4°C overnight. After additional TBST washes, membranes were incubated with corresponding horseradish peroxidase-conjugated secondary antibodies (Bio-Rad) for 1 h at room temperature and detected by the enhanced chemiluminescence method (SuperSignal West Pico substrate; Pierce; Rockford, IL, USA).
Small interfering RNA transfection
A549 and A549/DTX cells were seeded in six-well plates and transfected at 60% confluency with siRNA duplexes against human Akt (100 nM), mTOR (100 nM), Beclin1 (60 nM), Atg5 (60 nM), or control siRNA by lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol. After 6 h incubation, the transfected cells were exposed to arenobufagin for various time points, followed by analysis of cell viability by MTT assay, of apoptosis by flow cytometry, or of autophagy by MDC staining assay. Transfection efficiency was determined to be more than 75% by fluorescein-labeled non-targeted siRNA control (Ambion).
Tumor xenograft model
All procedures and experiments involving animals in this study were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals. The study protocol was approved by the Animal Ethics Committee at Shanghai Jiao Tong University School of Medicine, PR China. Female athymic nude mice aged 4–5 weeks were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, PR China) and were housed in the Animal Resource Facility. Exponentially growing A549/DTX cells (5×106 in 100 µL PBS) were injected subcutaneously in the right flank of each mouse. Tumor xenografts were allowed to grow to an average size of 50–150 mm3 and were randomly assigned to three different treatment groups (six mice per group): (1) vehicle control (1% DMSO in physiological saline); (2) PL 20 mg/kg; (3) PL 60 mg/kg. The mice were administered PL via intraperitoneal (i.p.) injections for 4 weeks (5 days/week). Tumor size was measured on two axes with the aid of Vernier calipers and tumor volume (mm3) was calculated using the formula: 1/2(L × W2) where L is the longest and W is the shortest axis. Mice were euthanized at the end of the study and/or when tumor size exceeded 2000 mm3.
TUNEL assay for apoptotic cells
The type of cell death (necrosis/apoptosis) was evaluated by the terminal deoxynucleotidyl transferase-mediated deoxyuridine biotin nick-end labeling (TUNEL) assay with an Apo-Direct kit (Pharmingen, San Diego, CA, USA) as previously reported. 22 Briefly, after antigen retrieval, the tumor sections (4 µm-thick) were fixed by incubation with 4% paraformaldehyde at 4°C. The permeabilized sections were incubated with terminal deoxynucleotidyl transferase recombinant (rTdT) enzyme-catalysed reaction and nucleotide mixture for 60 min at 37°C in the dark. After immersion in stop/wash buffer for 15 min at room temperature, the sections were washed with PBS to remove unincorporated fluorescein-12-dUTP and the nuclei counterstained with hematoxylin.
Immunohistochemical detection of Ki-67-positive, p-Akt-positive, and p-mTOR-positive cells
Immunohistochemistry assays were performed using antibodies against Ki67, p-Akt and p-mTOR. After fixation, tumor sections (4 µm thick) were deparaffinized and rehydrated. Following rehydration, antigen retrieval was carried out by placing the slides in 10 mmol/L sodium citrate buffer (pH 6.0) at 95°C for 20 min followed by 20-min cooling. The sections were then washed in PBS and non-specific binding sites were blocked with 1% bovine serum albumin with 2% goat serum in PBS before incubation with antibody. After washing, the sections were incubated with biotinylated secondary antibody followed by horseradish peroxidase-conjugated streptavidin. The sections were further incubated with 2,4-diaminobenzidine substrate and counterstained with hematoxylin. Each sample was examined separately and scored by two pathologists.
Statistical analysis
Data are presented as mean ± SD unless otherwise indicated. The statistical significance of the difference between the values of control and treatment groups was determined by either Student t test or simple one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons using Prism version 5 (GraphPad Software, Inc.). Values of P <0.05 were considered statistically significant.
Results
The effect of PL on cell growth was investigated using MTT assay on human lung cancer A549 cells and corresponding docetaxel-resistant A549/DTX cells. As shown in Figure 1a, PL potently inhibited the growth of both A549 and A549/DTX cells in a dose- and time-dependent manner. Of note, both A549 and A549/DTX cells were sensitive to PL reflected by the results of cell viability assay. Colony formation assay further confirmed that PL inhibited the lung cancer cells growth in a dose-dependent manner (Figure 1b).

Effects of piperlongumine (PL) on lung cancer cell growth and apoptosis. (a) A549 and A549/DTX cells were treated with 0, 1, 3, 6, or 10 μM piperlongumine for 24, 48, or 72 h, cell viability was measured by MTT assay. Absorbance was read at 490 nm with averages from triplicate wells. (b) Effects of PL on colony formation. Two days after treatment at the indicated concentrations of PL, cells were cultured for another 10 days, and colonies counted. (c) A549 and A549/DTX cells were treated with different concentrations of PL (0, 6, and 10 μM) for 48 h, followed by Annexin V-FITC/PI staining. (d) A549 and A549/DTX cells were cultured in DMEM medium with 10% fetal bovine serum and treated with PL (0, 6, and 10 μM) for 48 h, whole-cell lysates were collected and subjected to western blot analysis with the indicated antibodies. Data are presented as mean ± SD (n = 3) from three independent experiments. * P <0.05, ** P <0.01 compared with the control group.
Annexin V-FITC and PI staining were used to examine whether the reduction in cell growth in A549 and A549/DTX cells by PL treatment was related to the induction of apoptosis, the lung cancer cells were treated with varying concentrations of PL and the percentage of apoptotic cells were assessed. As shown in Figure 1c, treatment of A549 and A549/DTX with PL for 48 h resulted in a dose-dependent increase in the number of apoptotic cells in both cell lines. Interestingly, A549/DTX cells showed lower sensitivity to PL-mediated apoptosis than A549 cells.
To further investigate the underlying mechanisms involved in PL-induced effects on apoptosis in A549 and A549/DTX cells, we determined the expression of several key cellular proteins involved in apoptosis. As shown in Figure 1d, treatment of A549 and A549/DTX cells with PL (6 and 10 µM) for 48 h resulted in a dose-dependent induction of cleavage PARP. In addition, Bcl-2 level was decreased, whereas Bax level was increased.
Because A549/DTX cells showed lower sensitivity to PL-mediated apoptosis than A549 cells, we examined whether PL induced autophagy, which would have influenced the sensitivity of cancer cells to chemotherapy-induced apoptosis. After treatment with PL, Cyto-ID Green reagent staining showed the relative fluorescence intensity of cells was significantly increased, indicating the occurrence of autophagy (Figure 2a). Similar data were achieved through MDC staining (Figure 2b). Lipidation of microtubule-associated protein 1 LC3, as an autophagy marker, coats autophagosomes during autophagy and is converted to LC3-II resulting in the appearance of the delayed electrophoretic mobility in gel. Treatment with PL (0, 3, 6, and 10 μM) for 48 h, led to a significant increase in LC3-II (Figure 2c). This increase was further enhanced by the pretreatment of the cells with pepstatin A, a protease inhibitor, which inhibits LC3-II turnover (Figure 2c). All these results supported the idea that PL induced autophagy in A549/DTX cells.
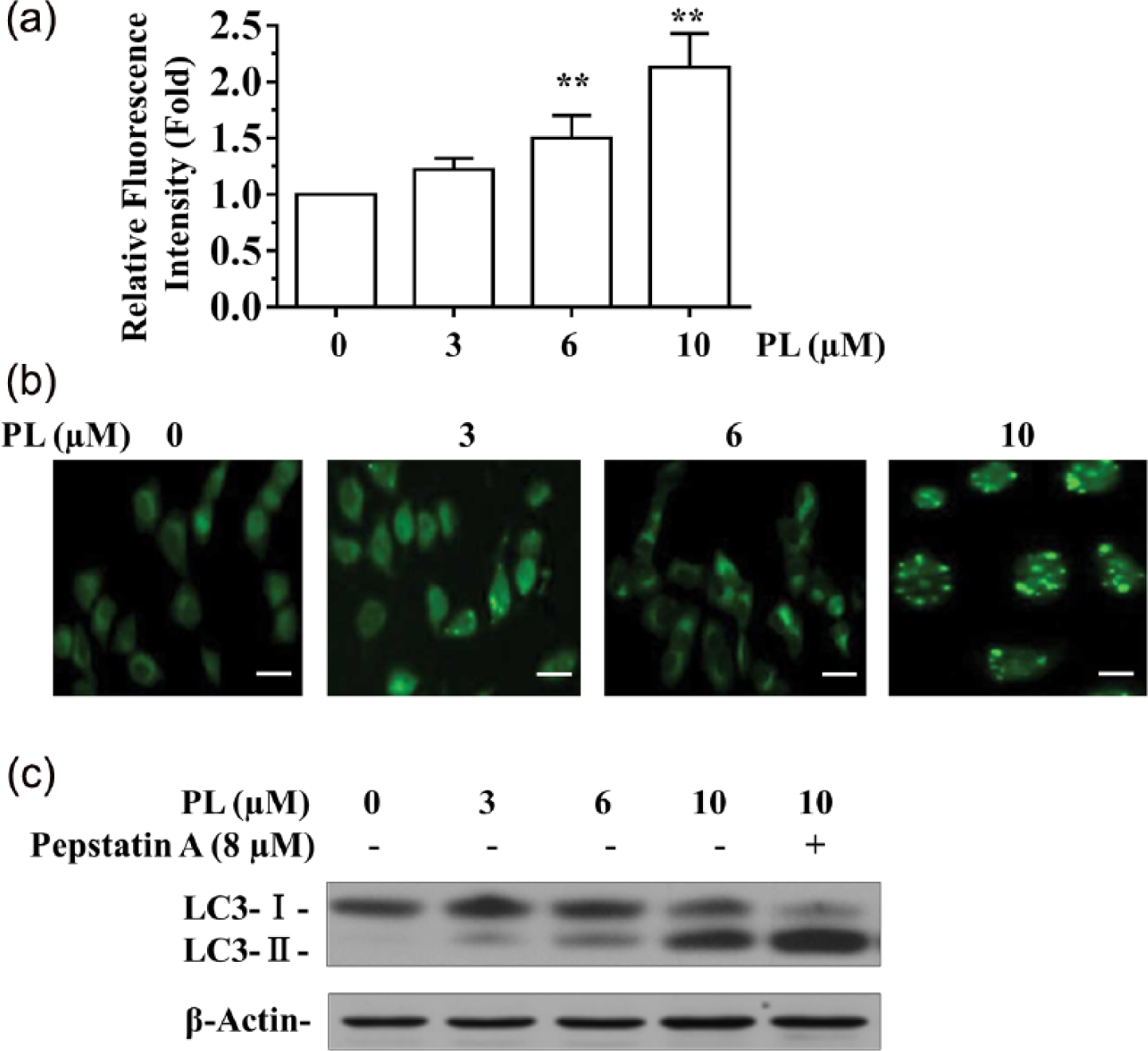
PL elicits autophagy in A549/DTX cells. (a) A549/DTX cells were incubated with PL (0, 3, 6, and 10 μM) for 48 h and stained with Cyto-ID for 30 min at 37°C. Intracellular Cyto-ID fluorescence was analyzed by Tecan GeNios microplate reader. (b) A549/DTX cells were incubated with PL (0, 3, 6, and 10 μM) for 48 h and stained with MDC. Intracellular MDC fluorescence was analyzed by confocal fluorescence microscopy. (c) Immunoblot analysis of LC3 in A549/DTX cells treated by PL and Pepstatin A (8 µM, 16 h). Data are presented as mean ± SD (n = 3) from three independent experiments. ** P <0.01 compared with the control group. Scale bars are 100 μm.
It has been reported that autophagy may facilitate cell survival in adverse microenvironment, and inhibition of autophagy may trigger increased induction of apoptosis in cells. 23 Recent studies suggested that autophagy may cooperate with apoptosis to promote cell death.24,25 We, therefore, explored whether inhibition of autophagy increased apoptosis in A549/DTX cells under PL treatment. This transition to apoptosis could be interrogated using both pharmacological and genetic inhibitors of different points of the autophagic pathway and assessing the induction of apoptosis. Here, we employed 3-methyladenine (3-MA), a specific autophagy inhibitor to target the earliest stages of autophagosome formation. Alternatively, inhibition of autophagy could be achieved by selective inhibition of Beclin1 and Atg5 using RNA interference. Both Beclin1 and Atg5 are common targets for autophagy inhibition. Pretreatment with 3-MA remarkably attenuated the formation of acidic autophagic vacuoles in the presence of PL (Figure 3a) and blocked the appearance of LC3-II in gel (Figure 3b). Addition of 3-MA also significantly increased the proportion of apoptotic cells when compared with those with PL alone (Figure 3c). Similar phenomena were observed in cells after genetic knockdown of the essential autophagy genes Beclin1/Atg5 (Figure 3c). In summary, these results suggested that autophagy may protect cells from PL-induced apoptotic cell death, and blocking autophagy by pharmacologic or molecular inhibition improved the killing effect of PL in A549/DTX cells through increased apoptosis.
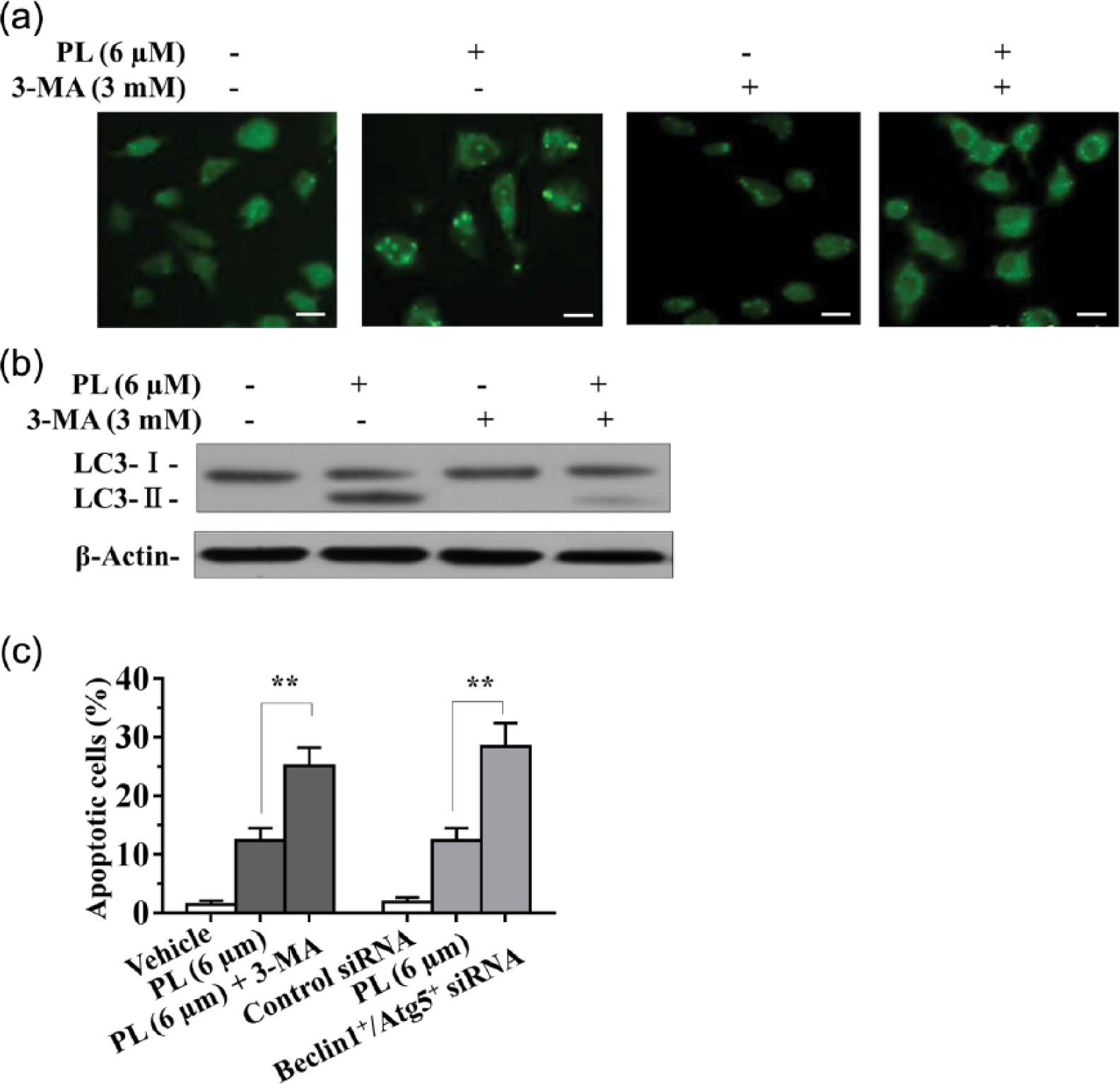
Inhibition of autophagy enhances apoptosis in PL-treated A549/DTX cells. (a, b) A549/DTX cells were pretreated with 3-MA (3 mM) for 30 min prior to a 48 h treatment with PL (6 µM). Cells were stained with MDC (a), or LC3 was analyzed by western blot (b). (c) A549/DTX cells treated with the combination of PL (6 µM) and 3-MA (3 mM) or Beclin+/Atg5+ siRNA duplexes. Cell apoptosis was analyzed by Annexin V-FITC/PI staining (c). Data are presented as mean ± SD (n = 3) from three independent experiments. ** P <0.01 compared with the control group. Scale bars are 100 μm.
Given the critical role of PI3K/Akt/mTOR pathway in controlling cell survival/death in response to external stimuli, 26 we investigated whether this pathway plays a central role in PL-mediated cell death. As shown in Figure 4a, when A549 and A549/DTX cells were treated with various concentrations of PL (0, 3, 6, and 10 μM) for 48 h, the levels of PI3K, phosphorylation of Akt at Thr308 and Ser473, and its downstream factor phosphorylation of mTOR (Ser2448) were effectively suppressed in a concentration-dependent manner. To further identify the role of Akt and mTOR in PL-mediated cell growth inhibition, we employed the Akt inhibitor LY294002 and mTOR blockers or siRNA to silence Akt and mTOR, respectively. We then examined the impact of PL on cell viability. The results showed that inactivation of Akt and mTOR tremendously sensitized A549 and A549/DTX cells toward cytotoxicity of PL (Figure 4b). These results together revealed that PL inhibits PI3K/Akt/mTOR signaling pathway in lung cancer cells.

PL inhibits the aberrant activation of PI3K/Akt/mTOR pathway. (a) A549 and A549/DTX cells were treated with PL (0, 3, 6, and 10 μM) for 48 h, and expressions of PI3K, Akt, p-AktThr308, p-AktSer473, phosphorylation of mTOR (Ser2448) in total cell lysates were evaluated by western blot analysis. (b) A549 and A549/DTX cells were treated with PL (3 μM) in the absence or presence of LY294002 (4 μM) and Akt siRNA (100 nM), or treated with mTOR blocker rapamycin (50 μM) for 30 min or transfected with mTOR siRNA (100 nM) for 6 h prior to 48 h treatment with PL. Cell viability was measured by MTT assay. Data are presented as mean ± SD (n = 3) from three independent experiments. ** P <0.01 compared with the PL alone group.
As these in vitro studies indicated that treatment of A549 and A549/DTX cells with PL reduced the cell growth and induces apoptosis of these cells, we sought to determine whether administration of PL inhibits in vivo tumor growth using a A549/DTX xenograft mouse model. As seen in Figure 5a, PL significantly suppressed tumor growth at doses of 20 and 60 mg/kg for 28 days compared with the control group. PL administration resulted in significant reduction of tumor volume in the nude mice. Animal weight loss is commonly used as a surrogate marker of toxicity. The average body weights of the PL-treated and non-PL-treated mice were comparable throughout the experimental period. As seen in Figure 5b, there was no difference in body weight and any other abnormality in food intake or behavior in PL treatment groups compared with the control group. These data suggested that administration of PL at the concentration used in these studies was not associated with apparent gross toxicity.

PL inhibits in vivo tumor xenograft growth in athymic nude mice. (a) Anti-tumor activity of PL in nude mice bearing A549/DTX tumors. Vehicle control and PL (20 and 60 mg/kg, i.p.) were administered for 4 weeks (5 days/week). (b) Effect of PL on body weight. (c) Tumors were excised and processed for immunostaining for Ki-67, TUNEL, p-Akt, and p-mTOR. Scale bars are 100 μm. Data in the graphs represent the mean ± SD (6 mice per group). **P <0.01, versus vehicle control.
Uncontrolled tumor cell proliferation is a characteristic feature of most cancers. We therefore analyzed the A549/DTX tumor xenografts for the potential anti-proliferative effects of PL using immunohistochemical detection of Ki-67-positive cells. As shown in Figure 5c, PL decreased the expression of Ki-67, a cell proliferation marker. In addition, to determine whether inhibition of tumor growth by administration PL is caused by the apoptosis of tumor cells in xenograft tissues, the apoptotic effect of PL on A549/DTX tumor tissues was identified by expression of the DNA fragment by TUNEL assay. The results showed that greater numbers of TUNEL-positive cells in the samples from PL-treated as compared with the control group. Furthermore, we determined the effect of PL on PI3K/Akt/mTOR pathway in tumor xenograft samples. Immunohistochemical analysis suggested that administration PL resulted in decreased the phosphorylation of Akt and mTOR in the tumor xenografts compared with the non-PL-treated group.
Discussion
Cancer cells evade programmed cell death to support malignant growth.27,28 Therefore, understanding the mechanisms of programmed cell death and designing therapeutic approaches to trigger cell death in cancer cells are critical for effectively treating the disease. The treatment of lung cancer remains a major challenge because of poor efficacy and severe toxicities of standard and new chemotherapy. There is an increased interest in seeking new therapies for lung cancers from natural compounds. Natural products have played an important role as an effective source of anti-tumor agents. It is estimated that up to 30–40% of the anti-cancer drugs used globally are derived from plant sources. 29 PL, a natural alkaloid present in the fruit of the Long pepper, is a promising bioactive molecule that has demonstrated anti-carcinogenic effects in some tumor models.19,20,30,31 Moreover, it has been reported that in a melanoma allografted mouse model, the anti-tumor effect of PL was greater than that of the cytotoxic chemotherapeutic drug, cisplatin. Similarly, PL outperformed paclitaxel in controlling a polyoma middle T antigen-driven mouse model of spontaneous mammary tumorigenesis. 32 However, the role of PL in lung cancer remains unclear. We therefore undertook a comprehensive analysis of the effects of PL on lung cancer cells using both in vitro and in vivo models. In this study, we demonstrated that PL was effective against drug-sensitive A549 cells as well as docetaxel-resistant A549/DTX cells. Its anti-tumor activity in vivo without weight loss or other life-threatening toxicities in animals supports its potential for further clinical investigation for lung cancer treatment. Our data also showed that PL induces apoptosis in A549 and A549/DTX cells, accompanied by an increase of Bax/Bcl-2 expression ratio and cleavage of PARP. In accord with our findings, some researchers reported that PL induces apoptosis in human triple-negative breast cancer cells and ovarian cancer cells.33,34
It is intriguing that the apoptotic rate in A549/DTX cells was much lower than that of A549 cells suggesting the presence of a cytoprotective mechanism. We thus examined whether PL induced autophagy. Autophagy is a catabolic process in which cells respond to various stress stimuli, such as hypoxia, nutrient starvation, and DNA damage. 35 In this process, proteins or organelles, sequestered by double-membrane structures, fuse with lysosomes and are subsequently degraded by lysosomal hydrolases to be recycled to sustain metabolism. 36 However, the exact role of autophagy in cancer treatment, and whether it protects cells from cytotoxic effects of anti-cancer drugs by blocking apoptosis or kills cells as an alternate pathway of cell death is still controversial.12,37 Our data revealed that PL could induce autophagy accompanied by apoptosis in A549/DTX cells. We found that inhibition of autophagy by specific inhibitors (3-MA) or Beclin1+/Atg5+ siRNA markedly increased apoptosis. Collectively, these results indicate that autophagy might provide a protective mechanism against PL-induced apoptosis.
Increasing evidence indicates that the cross-talk between autophagy and apoptosis is made especially complicated by the fact that they share many common regulatory molecules, such as p53, Bcl-2, and the PI3K/Akt/mTOR signaling pathway.38,39 It is well known that the PI3K/Akt/mTOR pathway plays an important role in cell growth, survival, differentiation, and metabolism. 40 Inhibition of PI3K/Akt/mTOR signaling pathway causes cell death associated with apoptosis and/or autophagy.41,42 The present results indicated that PL regulates PI3K, Akt, and mTOR. Either Akt-specific inhibitor LY294002 or Akt gene silencing by siRNA pretreatment caused the decrease in cell viability. Furthermore, mTOR inhibitor rapamycin or mTOR siRNA pretreatment enhances the cytotoxicity of PL, which further proved the critical role of PI3K/Akt/mTOR pathway.
Although in vitro cell culture models are a good system for preliminary screening of the effects of chemotherapeutic agents; the observations must be verified in vivo using animal models prior to their potential consideration of their use in humans. We therefore used an in vivo model of xenografts of A549/DTX tumor cells in athymic nude mice to verify the chemotherapeutic potential of PL against lung cancer cell growth. Our study provided evidence that administration of PL inhibited the growth of A549/DTX lung tumor xenografts without any apparent sign of toxicity in the athymic nude mice. These data are in accordance with the decreased proliferation documented by Ki67 immunostaining. Additionally, PL induced apoptosis as indicated by TUNEL staining in tumor tissues, at least in part, by targeting PI3K/Akt/mTOR cell survival pathway. These in vivo results are consistent to our in vitro study.
In conclusion, the present study demonstrates that PL inhibits the PI3K/Akt/mTOR pathway and invokes strong anti-cancer activity against lung cancer by inducing apoptosis as well as autophagy. Thus PL appears to be an attractive bioactive phytochemical for lung cancer chemoprevention and/or treatment. Our studies provide a rationale for the development of PL as chemotherapeutic agent against lung cancer in the clinical setting. K-ras mutant lung tumors might benefit from PL. Further studies evaluating the anti-tumor effects of PL for normal and other K-ras mutant lung tumors are needed to advance this treatment approach to a clinical setting.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by Key Talent Project of Jiangsu Province (No. RC2011032), Major Projects of Hospital Management Center in Wuxi City (No. YGZX201212), and Project of National Science Foundation (No. 81401186).