
Abstract
A 34-year-old male developed bilateral recurrent pheochromocytomas 10 years after laparoscopic adrenal-sparing surgery for pheochromocytomas. Based on his clinical manifestations and germline REarranged during Transfection (RET) variant, the patient was ultimately diagnosed with multiple endocrine neoplasia type 2B. Based on drug susceptibility testing results from organoid-guided precision therapy, the patient underwent secondary adrenalectomy for the right lesion and received oral pralsetinib to control the left ones. Over 16 months of pralsetinib therapy, we found that the patient achieved sustained therapeutic benefits, specifically characterized by symptomatic relief, significant reduction in hormone levels, and shrinkage of the left adrenal masses. These findings indicate that pralsetinib is effective and safe for treating pheochromocytomas associated with RET missense mutation, but further clinical practices are warranted to confirm its efficacy and safety profile.
Background
Pheochromocytomas and paragangliomas (PPGLs) are rare neuroendocrine tumors, originating from adrenal medulla and extra-adrenal paraganglia, respectively, with an estimated annual incidence of 4–21 cases per 1,000,000 person-years. 1 Difference from the previously described definition, the 2022 WHO classification recognizes that all PPGLs possess metastatic potential analogous to that of epithelial neuroendocrine tumors. 2 Besides, PPGLs are one of the most heritable cancers with more than 20 susceptibility genes identified so far, and germline variants could be found in approximately 40% of cases. 3 Multiple endocrine neoplasia type 2 (MEN2) is one of the sources that contribute to hereditary PPGLs, possessing the mutations of REarranged during Transfection (RET), a proto-oncogene associated with encoding a transmembrane receptor of the tyrosine kinase family. Clinically, there are two subtypes: MEN2A and MEN2B. The former is more common (90%–95%) and consists of four variants 4 : (1) MEN2A, classical type, presented with medullary thyroid carcinoma (MTC) and pheochromocytoma and/or hyperparathyroidism; (2) MEN2A with cutaneous lichen amyloidosis; (3) MEN2A with Hirschsprung disease; and (4) MEN2A with familial MTC only. While the latter (5%–10%) mainly includes the presence of pheochromocytoma, MTC, and marfanoid habitus. Current guidelines suggest that genetic testing and counseling are recommended for all PPGLs, especially those young patients with more likely to have hereditary syndromes. 5 Although PPGLs are catecholamine-secreting tumors that may be complicated by severe cardiovascular conditions, therapeutic strategies for these masses are limited, and surgery is still the first option. However, the risk of tumor recurrence or distal metastases after the initial surgery ranges from 5% to 30% based on previous reports,6,7 arousing the awareness that precision medicine and long-term follow-up are necessary for PPGL patients. In addition, it remains unclear whether pralsetinib is effective for PPGLs with RET variants clinically.
In this report, we described a case of a 34-year-old male who presented with bilateral recurrent pheochromocytomas 10 years after receiving laparoscopic adrenal-sparing surgery, and was finally diagnosed with MEN2B based on his clinical performance and germline mutation. The patient received not only secondary open adrenalectomy for the right adrenal mass but also oral pralsetinib capsules for inhibiting the RET-related signal pathway to date. Combining surgery and targeted therapy, we found that the patient gained significant prognosis during the follow-up.
Case presentation
A 34-year-old male patient presented with headache, palpitations, and sweating 10 years ago. Thyroid ultrasound demonstrated multiple nodules, with the dominant lesions in the left and right lobes measuring 2.3 × 1.3 and 1.3 × 0.6 cm, respectively. Bilateral adrenal masses were also identified through abdominal CT, measuring approximately 4 × 4.9 cm (right) and 4.7 × 3.2 cm (left). In 2014, the patient underwent total thyroidectomy followed by bilateral adrenal-sparing surgery, and the postoperative pathology confirmed MTC and bilateral adrenal pheochromocytomas. The patient was subsequently initiated on levothyroxine replacement therapy at 175 μg daily without regular follow-up examinations.
In 2023, the patient experienced recurrent symptoms similar to those recorded a decade earlier (Table 1), and subsequent abdominal contrast enhancement CT showed that masses in the right and left adrenal glands measured 58 × 42 and 51 × 41 mm, respectively (Figure 1(a) and (b)). In addition, serum 3-methoxytyramine, metanephrine, normetanephrine, and calcitonin were markedly elevated (Table 2), demonstrating the recurrence of MTC and bilateral pheochromocytomas. Given the patient’s history of early-onset multifocal endocrine tumors and recurrences, germline genetic testing was performed, and we identified a heterozygous pathogenic RET proto-oncogene variant (c.2753T > C), confirming the diagnosis of MEN2 syndrome finally for this patient.
Collection of symptoms during the follow-up.
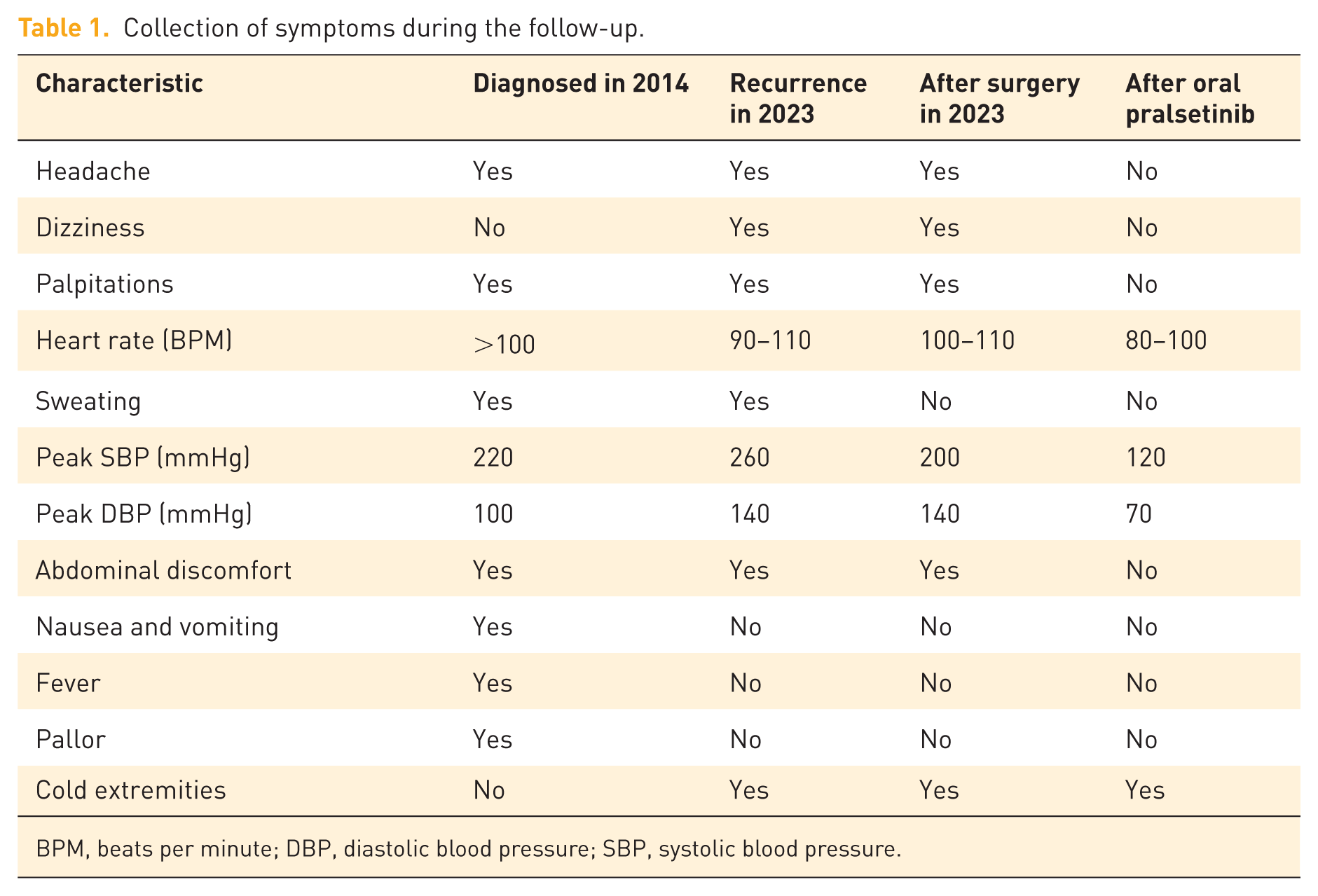
BPM, beats per minute; DBP, diastolic blood pressure; SBP, systolic blood pressure.

(a, b) Abdominal contrast enhancement CT done in 2023 showed bilateral adrenal masses, measuring 58 × 42 mm (85HU) and 51 × 41 mm (79HU) for the right and the left as shown by the red arrow, respectively, with another mass measuring 31 × 30 mm (109HU) in the left as shown by the yellow arrow. (c, d) Abdominal contrast enhancement CT done after 16-month pralsetinib therapy in 2025, the left adrenal mass measuring 35 × 32 mm (74HU) as shown by the blue arrow.
Changes in relevant hormones during the period of oral pralsetinib.

Detected 7 days after right adrenalectomy, as the baseline of oral pralsetinib.
3-MT, serum 3-methoxytyramine; CT, serum calcitonin; DA, serum dopamine; E, serum epinephrine; MN, serum metanephrine; NA, not available; NE, serum norepinephrine; NMN, serum normetanephrine.
After multidisciplinary evaluation, considering the patient’s young age and physiological need for adrenal relevant hormones, right adrenalectomy was performed, while the left adrenal gland (containing masses) was preserved. As expected, the immunohistochemistry (Figure 2) of the resected specimen confirmed recurrent pheochromocytoma, demonstrating chromogranin A (+), synaptophysin (+), and neuron-specific enolase (+).
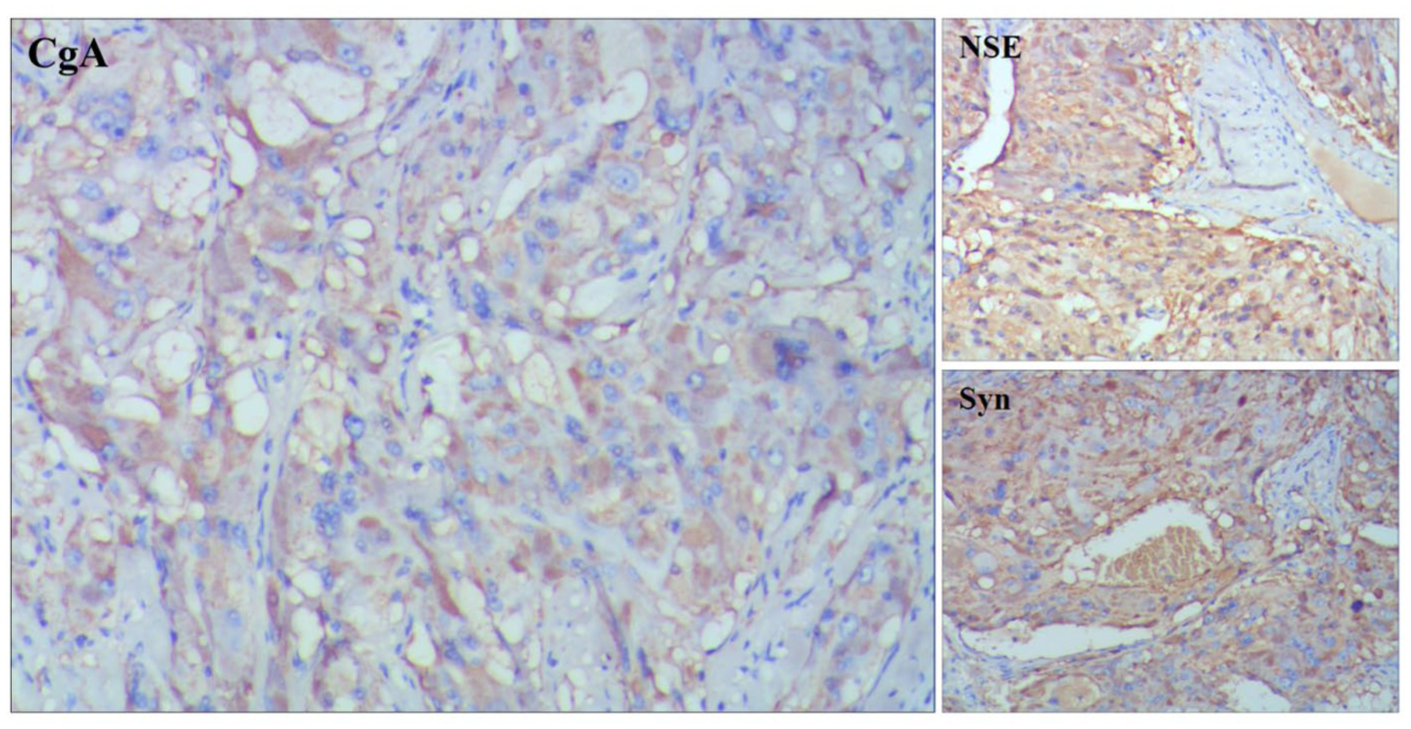
Immunohistochemistry of secondary surgery for right adrenal recurrent tumor in our hospital with positive CgA, NSE, and Syn.
For the left adrenal and thyroid lesions, the patient had taken orally pralsetinib (also called Gavreto, Blueprint Medicines, USA) 200 mg once daily since 1 month after postoperation for 7 months, and then reduced the dose to 100 mg once daily to date. Following 16 months of molecular targeted therapy, the recent adrenal contrast-enhanced CT reexamined in our hospital revealed two left adrenal lesions, with the largest one measuring 35 × 32 mm (Figure 1(c) and (d)), showing a reduction in volumes compared to baseline. In addition, there were non-enhancing regions within the tumor, suggestive of necrotic changes locally. In addition, he maintained clinical stability without symptoms related to the disease (Table 1), but experienced nausea and dizziness in the first month of oral pralsetinib, which improved subsequently. Serial biochemical monitoring demonstrated a marked decline in serum catecholamines and their metabolites, and calcitonin levels, though some values remained above reference ranges (Table 2).
Patient-derived organoid model
Prior to formulating the therapeutic regimen, we developed patient-derived organoid models using pheochromocytoma specimens from another MEN2 syndrome case harboring the identical RET mutation (Figure 3). Following our specific organoid culture protocols, systematic pharmacological evaluation of the selected five inhibitors was executed, and pralsetinib demonstrated superior tumor growth inhibition efficacy (59.07%) compared with selpercatinib (6.89%), cabozantinib (6.28%), surufatinib (3.14%), and vandetanib (0.18%; Figure 4).

Patient-derived organoid models of MEN2-related pheochromocytoma. (a) Day 1 (initial seeding phase). (b) Day 3 (organoid formation phase). (c) Day 12 (pre-treatment phase).
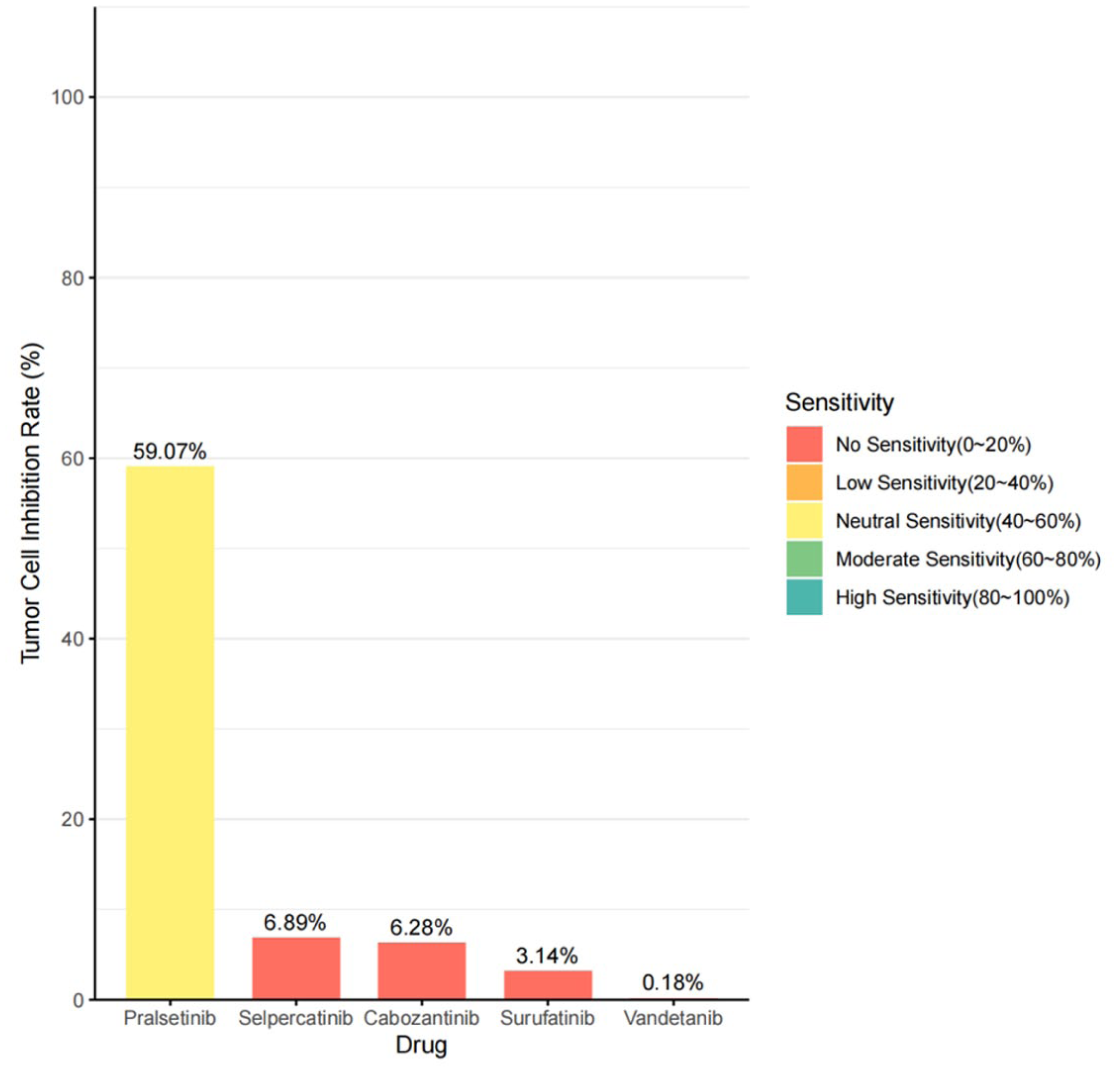
The inhibition rate of five drugs for tumor cells.
Discussion
Most PPGLs develop sporadically, while approximately one-third of cases are associated with clinically relevant syndromes, including von Hippel–Lindau disease, neurofibromatosis type 1, hereditary paraganglioma (caused by SDHx), familial pheochromocytoma (caused by TMEM127), and MEN2 syndrome (caused by RET). 8 Compared with sporadic PPGLs, hereditary ones are diagnosed at a younger age and tend to develop multiple tumors bilaterally. 5 In terms of treatment, current therapeutic options available for PPGLs have remained relatively stagnant, and it is urgent to develop novel therapeutic strategies, especially for those hereditary and metastatic ones. However, due to the significant heterogeneity of PPGLs, it is critical to conduct mutational detection and analysis in determining the optimal therapeutic strategy. Although some molecular and radionuclide therapies, such as tyrosine kinase inhibitors, HIF2α inhibitor (like Belzutifan), and peptide receptor radionuclide therapy, have been increasingly applied to treat PPGLs in clinical trials and show certain efficacy,9,10 no research focuses on selective RET-directed therapies for pheochromocytoma diagnosed with MEN2 syndrome to date.
The patient that we reported presented with bilateral pheochromocytomas and MTC simultaneously, and initially received bilateral adrenal-sparing surgery to avoid adrenal insufficiency at the age of 24. However, he presented with recurrent MTC as well as bilateral pheochromocytomas 10 years after the operation. The genetic screening tested in our hospital manifested that the heterozygous mutation of RET (c.2753T > C), resulting in amino acid 918 shifting from methionine (Met) to threonine (Thr), namely p.Met918Thr, which is a highly frequent mutation for the RET gene. The RET Met918Thr mutation is largely associated with an aggressive clinical course and poor prognosis in MTC patients, and approximately 95% patients with MEN2B possess this missense mutation. 4 Finally, the subtype of MEN2B was considered for this patient according to the clinical performance and mutation analysis. For MEN2 syndrome, most studies focused on MTC, as it represents the first manifestation of the disease, and can lead to a fatal outcome if undiagnosed or inappropriately treated. 11 While equal attention should be paid to MEN2-related pheochromocytoma, it is not only the second most frequent disease in patients with MEN2, but also recurs, metastasizes, and causes life-threatening cardiovascular complications. 12
RET alterations have been characterized as oncogenic drivers in multiple cancers, including MEN2-related MTC and pheochromocytoma, papillary thyroid carcinomas, non-small-cell lung cancer (NSCLC), and so on. Multikinase inhibitors (MKIs) with anti-RET activities have been used in advanced metastatic MTC and RET fusion-positive NSCLC, but their effectiveness is limited by obvious drawbacks, like off-target toxicities.13,14 Because of the limitations of MKIs, two highly selective RET inhibitors have been developed in recent years, pralsetinib and selpercatinib. 4 Pralsetinib, originally known as BLU-667 and now marketed as Gavreto, 15 was FDA approved in September 2020 for treating NSCLC harboring RET fusions, based on the multi-cohort clinical trial (ARROW, NCT03037385). 16 While, for MEN2 patients, recent clinical trials have demonstrated that pralsetinib was effective with acceptable safety to treat RET-mutant MTC.17,18 Although pralsetinib has been used to treat lung and thyroid cancers with RET mutations or fusions, and two recent case reports have demonstrated the efficacy of selpercatinib in treating pheochromocytomas with RET alterations,19,20 there is still a lack of clinical outcomes of pralsetinib in treating pheochromocytomas, especially those with MEN2.
We first reported the application of pralsetinib to treat MEN2-related pheochromocytoma. The patient initially received oral pralsetinib at a dose of 200 mg once daily for 7 months. Due to prominent chest tightness, the dosage was adjusted to 100 mg once daily, and treatment was continued, given the sustained therapeutic response. During the 16-month targeted therapy, the patient achieved improvement in symptoms and a gradual decrease in the levels of relevant hormones. We thought this therapeutic response could be attributed to dual factors: partly because the patient received adrenalectomy for the right lesion, and partly because of oral pralsetinib, which could actually control the left adrenal lesions, consistent with the presence of local necrosis within the tumor as seen on abdominal CT. At the same time, the patient experienced well-tolerated nausea and dizziness in the first month, and slightly decreased in white blood cell count as well as neutrophil count during follow-up, but did not present with physically pralsetinib-related adverse events, such as dry mouth, pneumonitis, diarrhea, headache, myalgia, taste disorder, and so on, as it reported in previous clinical trials.17,18,21 This may be attributed to individual well tolerance and the low dosage compared to that of clinical trials (recommended 400 mg once daily). Therefore, dosage reduction of pralsetinib from 200 to 100 mg achieved clinical symptom relief and a marked decrease in the levels of catecholamines and their metabolites in this patient, implying the efficacy of low-dose pralsetinib for treating MEN2-related pheochromocytoma. Notably, 36% of patients required dosage reduction during treatment, yet durable responses were consistently observed with low-dose pralsetinib. 22 In addition, previous research has demonstrated that dose reductions, guided by the occurrence of adverse events, can achieve an appropriate balance between therapeutic efficacy and safety. 23
Conclusion
Pralsetinib is effective and safe for our patient to treat MEN2-related pheochromocytoma. However, our result was based on a single case, and oral medication was given for about 16 months. Continuous follow-up and relevant clinical trials recruiting more subjects are necessary to further confirm the efficacy and safety of pralsetinib to treat pheochromocytomas in patients with MEN2 syndrome.
Supplemental Material
sj-pdf-1-tae-10.1177_20420188251415255 – Supplemental material for Efficacy and safety of pralsetinib in multiple endocrine neoplasia type 2-associated pheochromocytoma: a case report
Supplemental material, sj-pdf-1-tae-10.1177_20420188251415255 for Efficacy and safety of pralsetinib in multiple endocrine neoplasia type 2-associated pheochromocytoma: a case report by Shiwei Chen, He Wang, Qi Shen, Wencong Han, Kun Peng, Tai Kang, Zejin Ou and Zheng Zhang in Therapeutic Advances in Endocrinology and Metabolism
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.