
Abstract
Background:
The proteome is vital for discovering therapeutic targets. We conducted a proteome-wide Mendelian randomization (MR) analysis to identify potential Type 2 Diabetes (T2D) biomarkers and therapeutic targets.
Methods:
Data from deCODE Genetics (4907 proteins in 35,559 individuals) and the FinnGen study (65,085 T2D cases, 335,112 controls) were analyzed using inverse-variance weighted MR. Robustness was ensured through reverse MR and external cohort validation. Bayesian weighted MR further corroborated results. Additional analyses included protein-protein interaction (PPI) networks, pathway enrichment, druggability evaluation, and single-cell expression analysis.
Results:
Proteome-wide MR analysis identified 233 proteins associated with T2D risk. After adjusting for false discovery rate at 0.05, 15 proteins remained significant. Further reverse MR and validation using external cohorts confirmed that TPST2 and CHRDL1 (Chordin-like 1) were identified as the most promising potential therapeutic targets. For the 233 proteins associated with T2D risk, we conducted Gene Ontology enrichment and KEGG pathway enrichment analyses. These causal proteins were found to be involved in regulating inflammation and oxidative stress, atherosclerosis progression, and intracellular signaling mechanisms. A PPI network identified the top 10 hub genes: IGF1R, PPARGC1A, PDGFRB, ADIPOQ, IL15, BDNF, MET, SCARB2, KDR, and VWF. Drug enrichment analysis revealed that IGF1R, PPARGC1A, ADIPOQ, MET, and von Willebrand Factor (VWF) are targeted by metformin. Notably, sunitinib targets IGF1R, PDGFRB, MET, KDR, and VWF. Single-cell RNA sequencing confirmed these proteins’ expression.
Conclusion:
This study identifies novel T2D therapeutic targets and highlights sunitinib as a promising candidate. Future work should validate these findings and assess sunitinib’s efficacy in clinical trials.
Plain language summary
We used advanced methods to study proteins in the blood of people with Type 2 Diabetes. By analyzing these proteins, we found several that are linked to diabetes risk. We also discovered that a drug called sunitinib might be effective in treating diabetes because it targets some of these proteins. Our findings suggest that sunitinib could be a promising treatment option, but more research is needed to confirm this.
Introduction
Type 2 diabetes (T2D) has emerged as a critical public health issue globally. Its main features are insulin resistance and dysfunction of pancreatic beta cells, leading to prolonged hyperglycemia. 1 T2D not only significantly impacts patients’ quality of life but is also closely associated with severe health complications such as cardiovascular disease, stroke, renal failure, retinopathy, and neuropathy. These complications further reduce life expectancy for affected individuals. According to the most recent Global Burden of Disease Study from 2021, approximately 529 million adults worldwide have T2D, accounting for 6.1% of the global population (with a 95% uncertainty interval of 5.8%–6.5%). 2 It is projected that by 2050, this number will surpass 1.31 billion, imposing substantial pressure on public health systems and economies. 2 In addition, health issues related to T2D result in significant economic costs. In 2019, global direct medical expenditures reached $760 billion. 2 Notably, T2D has become the ninth leading cause of death worldwide, contributing to over 1 million deaths in 2017 alone, underscoring its profound impact on public health. 3
With the changing disease patterns, patients with T2D frequently experience comorbidities such as hypertension, cardiovascular diseases, and liver and kidney dysfunction. 4 These comorbidities not only accelerate the progression of T2D but also significantly complicate its treatment. 5 Although drugs such as metformin, SGLT2 inhibitors, and GLP-1 receptor agonists have been used to reduce the risk of complications or delay their onset in patients with T2D, these treatments are not always very effective for long-term blood sugar control and often come with side effects.6,7 Moreover, existing therapies typically require long-term medication, leading to frequent issues with patient compliance and failing to address the underlying pathophysiological mechanisms of T2D. 8 Therefore, discovering novel therapeutic targets and creating more effective treatments with reduced side effects is crucial for enhancing the prognosis of T2D patients.
In recent years, advancements in proteomics and genomics technologies have provided strong support for the study of plasma proteins as disease biomarkers and therapeutic targets. 9 Plasma proteins are involved in various biological processes (BPs), including metabolism, immunity, coagulation, and signal transduction, and are closely related to the onset and progression of T2D.10,11 Proteomics technologies offer new perspectives on the mechanisms underlying T2D development, with high-throughput screening potentially identifying serum markers that predict the occurrence and prognosis of T2D. This is significant for understanding the mechanisms of antidiabetic drugs, drug toxicology, and obtaining information on drug efficacy. 12 Zou and Yang 13 reported that they identified 72 potential protein targets for type 1 diabetes (T1D) and its complications, with MANSC4, CTRB1, SIGLEC5, and MST1 confirmed as key targets. This provides new insights and support for drug development and precision treatment of T1D. 13 Studies focusing solely on inflammatory proteins found that elevated levels of TGF-α and CX3CL1 are associated with the onset of T2D, while increases in FGF-21 and hGDNF reduce the risk of T2D. 14 However, there is still a lack of large-scale proteomics studies based on deCODE Genetics data (involving 35,559 individuals and 4907 proteins) specifically aimed at exploring risk factors and potential therapeutic targets for T2D. Therefore, there is an urgent need for more research to fill the gaps in comprehensive proteomics and provide new strategies and methods for the prevention and treatment of T2D.
Mendelian randomization (MR) utilizes naturally occurring genetic variations as tools in observational studies to explore the causal relationships between exposure factors and diseases, thereby minimizing reverse causality and bias. 15 Recent proteomics studies using this method have provided important insights into the prioritization of drug targets and have revealed potential causes of complex diseases such as cardiomyopathy, stroke, T1D, and hypertension.16–18 This approach not only enhances our understanding of the pathogenic mechanisms of these diseases but also lays the foundation for developing new therapeutic strategies.
In this study, we integrated comprehensive human plasma proteomics and genomic data to identify potential causal relationships between circulating protein biomarkers and T2D phenotypes. Utilizing Bayesian weighted MR (BWMR) analysis and incorporating two independent genetic datasets for validation, our primary objective was to screen for high-potential drug targets and predict their interactions with existing therapeutic agents. To further validate these biomarkers and therapeutic targets, we employed single-cell RNA sequencing (scRNA-seq) to investigate their expression patterns in specific cell types within T2D islet tissues. This approach aims to enhance our understanding of T2D pathogenic mechanisms and establish a robust foundation for developing novel therapeutic strategies.
Methods
All data used in this study were sourced from published research or public databases with informed consent and ethical approval. No additional ethical review was required. We confirm adherence to standard reporting guidelines and compliance with relevant ethical standards, ensuring the protection of participant rights and data legality.
Design
The study design is illustrated in Figure 1. Our primary aim was to evaluate the causal relationships between plasma proteins and T2D, and to explore their potential as therapeutic targets. We first performed a two-sample MR analysis using summary-level data from large-scale proteomic and genetic studies. To control for multiple testing, we applied FDR correction (FDR <0.05) and considered proteins with nominal significance but not passing FDR as suggestive findings. To strengthen causal inference, we conducted reverse MR to assess potential reverse causality and applied BWMR to account for possible pleiotropy among genetic instruments (Supplemental Material).

Flowchart depicting the step-by-step study design and methodology.
For proteins showing suggestive or significant associations, we performed downstream functional analyses to explore their biological relevance. This included Gene Ontology (GO) and KEGG pathway enrichment analyses to identify overrepresented BPs and signaling pathways. We further constructed protein-protein interaction (PPI) networks using STRING and Cytoscape to identify key hub genes. Candidate proteins were then evaluated for druggability through drug enrichment analyses using public databases such as DrugBank and DGIdb, followed by molecular docking simulations to assess the binding affinity of potential therapeutic compounds. Finally, we used scRNA-seq data to validate the expression of key targets in disease-relevant cell types, thereby strengthening their biological plausibility as therapeutic candidates.
Proteomic data source
This study utilized data from the deCODE project’s pqtl dataset, which encompasses genetic information for 4907 proteins measured in 35,559 Icelandic individuals. 19 The dataset provides extensive protein expression variation, ensuring both breadth and reliability of our analysis. To ensure high-quality data, we implemented several preprocessing and quality control steps. First, we harmonized genetic variants across different datasets using the TwoSampleMR package in R (version 4.4.1), ensuring consistency in allele coding and strand orientation. Variants with minor allele frequency <0.01 or failing Hardy-Weinberg Equilibrium tests (p < 1 × 10⁻⁶) were excluded to minimize potential biases. In addition, linkage disequilibrium (LD) pruning was performed using an R2 threshold of 0.001 to reduce collinearity among SNPs. Using this large-scale dataset, we were able to accurately identify biomarkers and therapeutic targets associated with T2D. Specifically, the comprehensive coverage of protein expression levels allowed us to detect subtle variations that might be missed in smaller studies. This robust dataset provided a solid foundation for developing new T2D treatment strategies by enabling precise identification of proteins involved in disease mechanisms and their potential as therapeutic targets.
Outcome data sources for MR
T2D outcome data were obtained from the FinnGen R10 dataset, which includes 65,085 T2D cases and 335,112 controls of European ancestry (https://storage.googleapis.com/finngen-public-data-r10/summary_stats/finngen_R10_T2D.gz). FinnGen is a large-scale biobank-based study combining genotype data with national health registries, providing high-quality, well-phenotyped case-control data. This dataset was selected as the primary source due to its large sample size, population homogeneity, and rigorous phenotyping standards.
For replication and cross-population validation, we used two independent GWAS datasets. The first was from a European-ancestry cohort (EBI accession: ebi-a-GCST006867), comprising 61,714 T2D cases and 655,666 individuals with 5,030,727 SNPs. The second was from an East Asian cohort (BBJ accession: bbj-a-153), including 40,250 cases and 170,615 controls, with a total of 210,865 individuals and 8,885,694 SNPs. These datasets were selected to ensure robustness of findings across different ethnic populations and to minimize potential biases due to population stratification. All datasets are publicly available, extensively quality-controlled, and have been widely used in genetic studies of T2D, further supporting the validity of our MR analyses.
MR analysis and BWMR analysis
We conducted MR analyses to evaluate the causal relationship between plasma protein levels and T2D risk. Genetic instrumental variables (IVs) were selected based on genome-wide significance (p < 5 × 10−8) from proteomic quantitative trait locus data. To minimize LD, we applied an R2 threshold of <0.001 using the clump_data function in the TwoSampleMR R package (version 4.4.1). 20 The strength of each IV was assessed using the F-statistic, calculated as F = β 2 /SE 2 , where β is the effect size and SE is the standard error. 21 Instruments with F > 10 were considered to have minimal weak instrument bias.
To comprehensively evaluate causality, we applied five MR methods: inverse-variance weighted (IVW), weighted median, MR-Egger, weighted mode, and simple mode. These methods provide complementary insights into the causal estimates and help detect potential violations of MR assumptions. We tested for heterogeneity using the IVW Cochran’s Q test and assessed horizontal pleiotropy using MR-PRESSO global and outlier tests. 22 The analysis adhered to the three core MR assumptions: (1) relevance (IVs are associated with the exposure), (2) independence (IVs are not associated with confounders), and (3) exclusion restriction (IVs affect the outcome only through the exposure). 13
To further improve the robustness of our findings and reduce the influence of pleiotropy, we applied BWMR analysis. In BWMR, each genetic instrument is assigned a weight based on its posterior probability of being a valid IV, thereby down-weighting potentially pleiotropic variants. This approach was implemented using the BWMR R package, with default priors and convergence criteria. Finally, all significant findings were validated in two independent cohorts: a European cohort (EBI accession: ebi-a-GCST006867, n = 655,666) and an East Asian cohort (BBJ accession: bbj-a-153, n = 210,865), ensuring cross-population consistency and strengthening the validity of our causal inferences.
GO and KEGG pathway enrichment analysis
To explore the biological functions and signaling pathways associated with the identified plasma proteins, we conducted GO and KEGG pathway enrichment analyses. GO analysis was performed across three functional categories: BP, molecular function (MF), and cellular component (CC), enabling a multilevel understanding of the biological roles of candidate genes. 23 KEGG pathway analysis provided integrated insights into gene-disease associations, signaling pathways, and potential drug targets, supporting the identification of pathophysiological mechanisms related to T2D. 24
All enrichment analyses were carried out using the clusterProfiler R package (version 4.2.0), 25 with the org.Hs.eg.db annotation package used for gene ID mapping. Candidate genes were input as a gene list, and enrichment was tested using the hypergeometric test, with significance set at p < 0.05 after Benjamini–Hochberg correction for multiple testing. Enriched pathways with a minimum of five mapped genes and an FDR <0.05 were considered statistically significant. To enhance interpretability, we visualized the top enriched pathways using bubble plots generated with the ggplot2 package. These functional annotations provided insights into the molecular mechanisms underlying the identified protein–T2D associations and helped prioritize potential therapeutic targets for further investigation.
PPI and druggability evaluation with molecular docking
To explore the functional interactions among the identified candidate genes and identify key regulatory hubs, we performed PPI network analysis using the STRING database (version 11.5; https://string-db.org/) and visualized the results using Cytoscape (version 3.10.3). STRING was used to retrieve both direct physical and indirect functional associations between proteins, with a minimum interaction confidence score of 0.4 set as the threshold for inclusion. The resulting network was further analyzed in Cytoscape to identify the top 10 hub genes based on node connectivity using the CytoHubba plugin. These hub genes, characterized by high connectivity, were considered central to the underlying BPs and prioritized for further functional and therapeutic evaluation.
To assess the druggability of these hub proteins, we conducted drug enrichment analysis using multiple public databases, including DrugBank (https://go.drugbank.com/), 26 DGIdb (https://www.dgidb.org/), 27 and ChEMBL (https://www.ebi.ac.uk/chembl/). 28 These databases provide comprehensive information on known drug-target interactions, enabling us to identify existing or investigational drugs that potentially target the hub proteins. Candidate targets with documented interactions in at least two of these databases were considered as promising druggable targets.
To further validate the interaction potential between selected ligands and core target proteins, we performed molecular docking simulations using CB-Dock2 (https://cadd.labshare.cn/cb-dock2/index.php). This tool enables accurate estimation of binding affinity and docking poses between small molecules and protein targets. Ligands with the lowest binding energy and favorable docking scores were prioritized as potential therapeutic candidates. These integrated analyses provided a robust framework for identifying and validating key proteins involved in T2D pathogenesis and evaluating their potential as therapeutic targets.
Single-cell-type expression analysis
To investigate the cell-type-specific expression patterns of the identified protein-coding genes, we obtained scRNA-seq data from the PanglaoDB database (https://panglaodb.se/), which includes a total of 1368 scRNA-seq datasets. We selected islet tissue samples (SRA701877: SRS3279692) for downstream analysis. The raw data were normalized and preprocessed using the Seurat R package, 29 and cell types were annotated using the SingleR package 30 based on reference transcriptomic profiles. To identify genes specifically enriched in certain cell types, we applied the FindAllMarkers function with thresholds of average log2 fold change >1 and p < 0.05. This approach enabled us to detect protein-coding genes that are differentially expressed across distinct cell populations within the pancreatic islets.
Statistical analysis
Statistical analyses were primarily conducted using two-sample MR and BWMR approaches. All MR analyses were performed with the TwoSampleMR R package (version 4.4.1), following standard pipelines for data harmonization, instrument selection, and causal effect estimation. For the binary outcome of T2D, we calculated odds ratios (ORs) and their corresponding 95% confidence intervals (CIs) to quantify the association between genetically predicted plasma protein levels and T2D risk. Causal estimates were considered statistically significant at a two-sided p-value <0.05 after Bonferroni or FDR correction for multiple comparisons, where applicable.
To account for potential pleiotropy and improve the robustness of causal inference, we applied BWMR analysis using the BWMR R package. This method assigns weights to genetic instruments based on their likelihood of being valid IVs, thereby reducing the influence of horizontal pleiotropy. Sensitivity analyses, including MR-Egger intercept tests and Cochran’s Q tests, were used to assess directional pleiotropy and heterogeneity, respectively.
Data visualization, including forest plots, scatter plots, and MR-Egger plots, was performed using R packages such as ggplot2 and forestplot. Final figure formatting was completed using Adobe Illustrator 2023 to ensure clarity and consistency in presentation. All statistical tests were two-sided, with a significance threshold of p < 0.05 unless otherwise specified.
Results
Initial screening of causal association between plasma proteins and T2D
To identify plasma proteins with potential causal effects on T2D, we performed a two-sample MR analysis on 4907 circulating proteins. Using the IVW method, we initially identified 240 proteins showing nominal association (p < 0.05). To improve confidence in causal inference, we applied a stricter criterion requiring consistent effect direction across all five MR methods (IVW, weighted median, MR-Egger, weighted mode, and simple mode), which narrowed the list to 233 proteins (Figures 2 and 3).

Graphical representations of MR analysis outcomes for circulating proteins. (a) Volcano plot. (b) Circular visualization. (c) Relationship between genetically predicted plasma protein levels and T2D risk, illustrated in a Manhattan plot.

Forest plot of MR estimates of the association between plasma proteins and type 2 diabetes. (a) Forward Mendelian randomization analysis. (b) Reverse Mendelian randomization analysis. (c) Bayesian weighted Mendelian randomization analysis.
After correcting for multiple testing using the FDR at 0.05, 15 proteins remained statistically significant. Among these, 6 showed protective associations with T2D risk, including THSD1 (OR = 0.915, 95% CI: 0.887–0.945, p = 3.45 × 10−⁸) and PCMT1 (OR = 0.739, 95% CI: 0.640–0.854, p = 3.82 × 10−⁵). The remaining four proteins—SUN5, FGFBP3, GDF5, and CHRDL1 (Chordin-like 1)—also demonstrated protective effects (all p < 0.0001).
Nine proteins were associated with increased T2D risk, with the strongest effects observed for HSPA1B (OR = 1.358, 95% CI: 1.207–1.528, p = 3.66 × 10−⁷) and GOLM1 (OR = 1.090, 95% CI: 1.052–1.130, p = 2.05 × 10−⁶). Other risk-associated proteins included TPST2, DIXDC1, GLCE, GALNT1, DCP1A, EXOSC8, and CD58 (all p < 0.0001). Sensitivity analyses, including Cochran’s Q test for heterogeneity and MR-Egger intercept for directional pleiotropy, did not reveal significant issues (all p > 0.05), indicating robustness of the observed associations.
BWMR analysis and reverse MR
To strengthen the reliability of our causal inferences, we applied BWMR to validate the 15 plasma proteins identified in the initial MR analysis (Figure 3). The BWMR results were highly consistent with those from the IVW method, supporting the robustness of the observed associations. Among the six proteins with protective effects, SUN5 showed the strongest association (OR = 0.843, 95% CI: 0.781–0.910, p = 1.22 × 10−⁵), followed by PCMT1 (OR = 0.737, p = 0.00014), CHRDL1 (OR = 0.772, p = 0.00014), GDF5 (OR = 0.884, p = 0.00016), FGFBP3 (OR = 0.917, p = 0.00016), and THSD1 (OR = 0.943, p = 0.0079). For the nine risk-increasing proteins, HSPA1B (OR = 1.362, p = 1.89 × 10−⁶) and TPST2 (OR = 1.229, p = 7.05 × 10−⁶) exhibited the strongest effects. Other significant associations included DIXDC1 (OR = 1.354, p = 4.34 × 10⁻⁵), GALNT1 (OR = 1.210, p = 7.52 × 10−⁵), DCP1A (OR = 1.093, p = 0.00021), EXOSC8 (OR = 1.205, p = 0.00025), CD58 (OR = 1.096, p = 0.0011), GLCE (OR = 1.076, p = 0.0038), and GOLM1 (OR = 1.064, p = 0.0073).
To further assess causality, we conducted reverse MR analysis to evaluate whether T2D influenced the levels of these proteins. Significant reverse associations were observed for GOLM1, TPST2, HSPA1B, GLCE, CHRDL1, and CD58, suggesting a bidirectional relationship between these proteins and T2D. These findings indicate that these proteins are not only associated with T2D risk but may also represent promising therapeutic targets (Figure 3).
External dataset validation
To validate the robustness of our findings, we evaluated the 15 identified proteins in two independent datasets: one from a European population and another from an Asian population. In the European dataset, three proteins—CHRDL1, GALNT1, and TPST2—showed significant associations with T2D. Similarly, in the Asian dataset, significant associations were observed for GOLM1, TPST2, and CHRDL1. Notably, TPST2 and CHRDL1 were consistently validated across both populations, highlighting their strong and reproducible associations with T2D. These findings support their potential as promising therapeutic targets for further investigation.
Enrichment analysis
To explore the biological functions and pathways associated with the 233 proteins showing initial causal associations with T2D, we performed GO and KEGG pathway enrichment analyses (Figure 4). GO analysis revealed significant enrichment across multiple BP, CC, and MF. Notable BP terms included “receptor-mediated endocytosis” and “cellular response to oxidative stress,” suggesting roles in metabolic regulation and stress response. Among CC terms, “collagen-containing extracellular matrix” was significantly enriched, indicating potential involvement in structural and tissue remodeling processes. In MF, “sulfur compound binding” and “glycosaminoglycan binding” were enriched, highlighting key roles in molecular interactions and signaling regulation.

Functional enrichment analysis of identified proteins using GO and KEGG pathways. (a) GO barplot. (b) GO bubble. (c) GO circular plot. (d) KEGG pathways barplot. (e) KEGG pathways bubble. (f) KEGG pathways circular plot.
KEGG pathway analysis identified several pathways significantly associated with T2D pathogenesis, including cytokine-cytokine receptor interaction, chemical carcinogenesis—reactive oxygen species (ROS), fluid shear stress and atherosclerosis, and the MAPK signaling pathway. These pathways are closely linked to inflammation, oxidative stress, insulin resistance, and atherosclerosis—key mechanisms in T2D development. They also involve multiple genes related to cell proliferation, apoptosis, and intracellular signaling, offering potential insights for drug-target discovery. Together, these findings provide a comprehensive overview of the molecular mechanisms underlying T2D and highlight several promising BPs and signaling pathways for further investigation.
PPI network analysis
To gain insights into the functional interactions and biological roles of the 15 proteins significantly associated with T2D, we conducted PPI network analysis. Using the GeneMANIA database, we first constructed an interaction network centered on the candidate proteins, including THSD1, PCMT1, CHRDL1, SUN5, GDF5, FGFBP3, GOLM1, GLCE, DCP1A, CD58, EXOSC8, GALNT1, TPST2, DIXDC1, and HSPA1B. GeneMANIA not only validated known interactions but also predicted additional functionally related genes, thereby expanding the network and enhancing our understanding of the potential roles of these proteins in T2D.
We further refined and expanded the network using the STRING database (version 12.0), incorporating experimentally validated interactions, text mining, and functional predictions. Only proteins with high-confidence interaction scores (⩾0.7) and p < 0.05 from MR analysis were included. The resulting network was visualized and analyzed using Cytoscape, from which we identified the top 10 hub genes: IGF1R, PPARGC1A, PDGFRB, ADIPOQ, IL15, BDNF, MET, SCARB2, KDR, and VWF (Figure 5). These hub genes are likely to play central roles in T2D pathogenesis, offering valuable candidates for functional studies and potential drug targets.
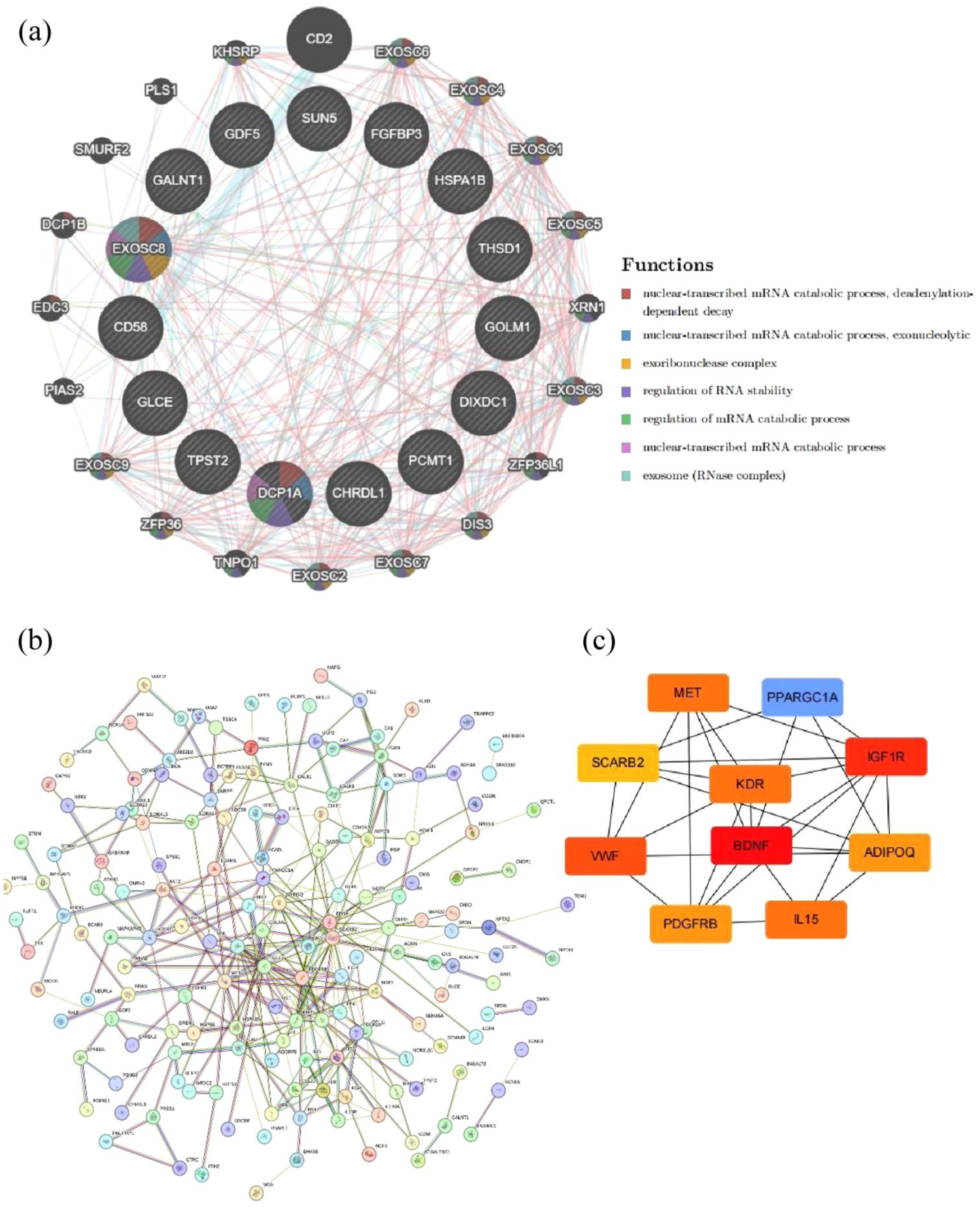
PPI analysis of identified proteins. (a) PPI network involving 15 proteins retrieved from the GeneMANIA database. (b) PPI results sourced from the STRING database and presented using Cytoscape Version 3.10.3. (c) PPI network of the top 10 hub genes from STRING.
Drug enrichment and molecular docking
To explore the therapeutic potential of the top 10 hub genes identified in the PPI network, we performed drug enrichment analysis (Figure 6). This analysis revealed significant interactions between metformin—a widely used T2D medication—and five hub genes: IGF1R, PPARGC1A, ADIPOQ, MET, and VWF, supporting the biological relevance of our network. Notably, sunitinib, a tyrosine kinase inhibitor, also showed significant interactions with five hub genes: IGF1R, PDGFRB, MET, KDR, and VWF, highlighting its potential as a novel candidate for T2D treatment.

Results of network pharmacology analysis. (a) Barplot. (b) Bubble chart. (c) Cnetplot.
To further validate these findings, we conducted molecular docking using an online platform (https://cadd.labshare.cn/cb-dock2/index.php). Based on minimized free energy, we selected the best docking conformations for sunitinib and its target proteins. Docking results confirmed strong binding affinities between sunitinib and IGF1R, PDGFRB, MET, KDR, and von Willebrand Factor (VWF; Figure 7). These results not only validate the known efficacy of metformin but also suggest that sunitinib may serve as a promising repurposed drug for T2D. This multilevel validation provides a solid foundation for future experimental and clinical investigations.

Docking patterns of the most promising drug molecules for five gene targets. (a) IGFR1. (b) KDR. (c) MET. (d) VWF. (e) PDGFRB.
Validation using scRNA-seq data
To evaluate whether the protein-coding genes associated with sunitinib and metformin are enriched in specific cell types in T2D, we performed scRNA-seq analysis on islet tissues from patients with T2D and healthy controls. This analysis identified 10 distinct cell types within the T2D group: β cells, ductal cells, α cells, acinar cells, pancreatic stellate cells, enteroendocrine cells, endothelial cells, δ cells, γ (PP) cells, and unclassified cells. All 10 hub protein-coding genes identified in our study were successfully validated in this dataset. Further investigation into the expression patterns of these genes revealed that IGF1R was predominantly expressed in β cells, ductal cells, and endothelial cells; PDGFRB was enriched in β cells and pancreatic stellate cells; MET showed significant expression in β cells and ductal cells; KDR and VWF were primarily enriched in endothelial cells; PPARGC1A was expressed in β cells and ductal cells; ADIPOQ was mainly found in β cells; IL15 was expressed in ductal cells, β cells, and pancreatic stellate cells; Brain-Derived Neurotrophic Factor (BDNF) was primarily detected in β cells; and SCARB2 was broadly present across all analyzed cell types. These findings not only confirm the cell-type-specific expression of key genes but also provide strong biological support for their relevance in T2D pathophysiology, highlighting their potential as cell-type-specific therapeutic targets. The results are summarized in Figure 8, which illustrates the expression levels of these hub genes across different cell types.
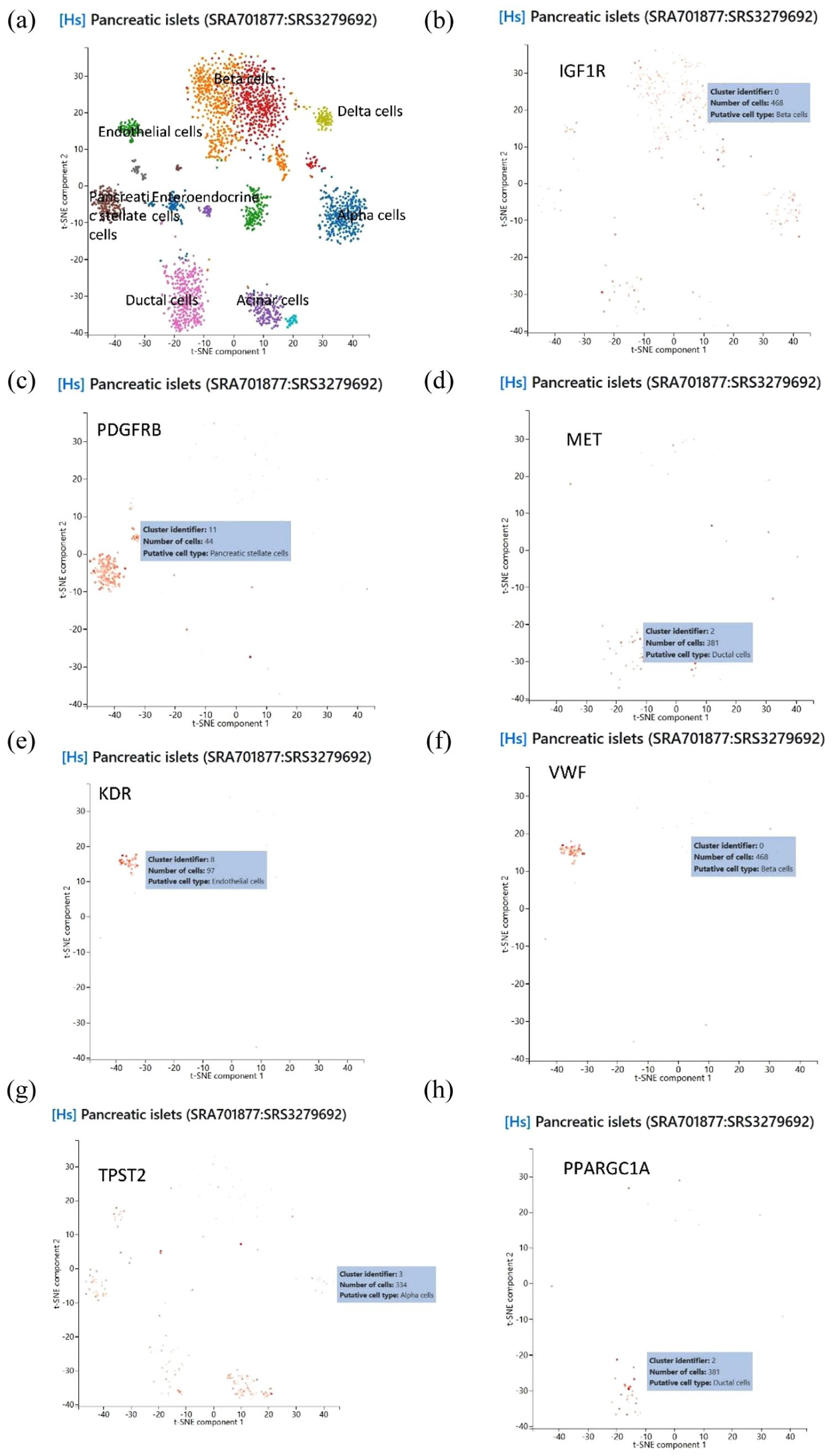
Results of single-cell sequencing analysis. (a) The total data of liver tissue from SRA701877:SRS3279692. (b) IGFR1. (c) PDGFRB. (d) MET. (e) KDR. (f) VWF. (g) TPST2. (h) PPARGC1A.
Discussion
This study employed a proteome-wide MR approach to investigate the causal relationships between 4907 circulating plasma proteins and T2D risk. Leveraging large-scale genetic and proteomic datasets, we identified 233 proteins nominally associated with T2D, of which 15 remained significant after multiple testing correction (FDR <0.05). Notably, five proteins (CHRDL1, PCMT1, SUN5, GDF5, and FGFBP3) showed protective effects, while 10 (including GOLM1, TPST2, and HSPA1B) were associated with increased T2D risk. Bidirectional MR analyses further revealed reverse causality for six proteins, suggesting potential feedback mechanisms in disease progression. Cross-population validation in European and East Asian cohorts confirmed TPST2 and CHRDL1 as the most robust candidates for further functional characterization. In parallel, network-based analyses identified a panel of hub genes—IGF1R, PPARGC1A, PDGFRB, ADIPOQ, and SCARB2, among others—implicated in key metabolic and inflammatory pathways. Importantly, molecular docking analyses identified sunitinib, a multitarget tyrosine kinase inhibitor, as a potential repurposable drug capable of interacting with several of these hub proteins, including IGF1R, PDGFRB, MET, KDR, and VWF. These findings provide novel insights into the molecular mechanisms underlying T2D and highlight promising therapeutic targets warranting further investigation.
Through drug-gene enrichment analysis, we identified metformin and sunitinib as the only two drugs targeting multiple hub genes associated with T2D, with three shared targets (IGF1R, MET, and VWF). This overlap not only supports the validity of our network-based approach but also suggests that sunitinib may share certain therapeutic mechanisms with metformin while offering broader multitarget coverage. Molecular docking analyses further confirmed strong binding affinities between sunitinib and key T2D-related proteins, including IGF1R, PDGFRB, MET, KDR, and VWF. These findings were supported by scRNA-seq data, which validated the expression of these targets in relevant metabolic tissues. Several experimental and clinical studies have demonstrated that sunitinib improves insulin sensitivity and β-cell function in diabetic models. For instance, sunitinib has been shown to enhance glucose-stimulated insulin secretion, reduce oxidative stress and inflammation, and improve endothelial dysfunction in T2D models.31–35 Notably, a case report described sunitinib-induced hypoglycemia during cancer treatment, further supporting its metabolic effects. 34 Together, these findings suggest that sunitinib, traditionally used as an anti-cancer agent, may have repurposing potential in T2D. However, its clinical application in metabolic diseases should be carefully evaluated in light of known resistance mechanisms in oncology.
Emerging evidence highlights the involvement of IGF1R, PDGFRB, KDR, MET, and VWF in the pathogenesis of T2D and its vascular complications, offering novel insights into potential therapeutic strategies. These genes are primarily implicated in key BPs such as insulin signaling, endothelial dysfunction, inflammation, and fibrosis—hallmarks of T2D and its complications. IGF1R (insulin-like growth factor 1 receptor) plays a central role in cell survival and proliferation and has been linked to insulin resistance and diabetic vascular complications. Studies have shown that sunitinib inhibits IGF1R activity, which may contribute to its potential insulin-sensitizing effects. 38 Notably, modulation of the IGF-1 signaling axis has been shown to ameliorate diabetic nephropathy in animal models,37,38 and a functional variant in IGF1R has been associated with increased T2D risk in specific populations. 39 PDGFRB (Platelet-Derived Growth Factor Receptor Beta) is a key regulator of angiogenesis and fibrosis, processes that underlie diabetic complications such as nephropathy, retinopathy, and atherosclerosis.40–42 Targeting the PDGF/PDGFR pathway has shown therapeutic promise in preclinical models, further supporting its relevance in T2D. KDR (VEGFR2), a major mediator of endothelial cell function, is dysregulated under hyperglycemic conditions. Its ligand-independent activation by ROS impairs VEGF signaling and contributes to defective angiogenesis in diabetes, 36 suggesting that VEGFR-targeted therapies such as sunitinib may help restore endothelial function. MET, also known as the hepatocyte growth factor receptor, plays a dual role in tumor progression and metabolic regulation. Studies have shown that MET enhances insulin signaling and improves glucose metabolism in insulin-resistant models,43,44 making it a promising target for T2D therapy. Given that sunitinib inhibits MET activity, this mechanism may contribute to its metabolic effects. Finally, VWF serves as a biomarker of endothelial dysfunction and thrombosis in T2D. Elevated VWF levels are associated with increased cardiovascular risk in diabetic patients,45–47 and their inhibition by sunitinib may help mitigate diabetes-related vascular complications. Together, these findings suggest that the five hub genes identified in our study are not only mechanistically relevant to T2D but also represent actionable therapeutic targets. Their shared involvement in metabolic and vascular pathways, along with their inhibition by sunitinib, supports further investigation of this multitarget kinase inhibitor in T2D.
Although our results suggest that sunitinib may have repurposing potential for the treatment of T2D, its drug resistance issues observed in other diseases warrant particular attention. Emerging evidence from oncology has revealed multiple mechanisms underlying sunitinib resistance, which may inform its future application in metabolic disorders. Ghosh et al. 48 found that sunitinib-resistant clear cell renal cell carcinoma (ccRCC) cells exhibit upregulated expression of Axl and PD-L1, along with metabolic reprogramming characterized by enhanced oxidative phosphorylation and glutamine metabolism. While inhibitors targeting Axl, AMPK, p38, or PD-L1 showed limited efficacy in co-culture models, the AMPK activator metformin enhanced PD-L1 blockade and improved cytotoxic T lymphocyte-mediated tumor killing. 48 These results suggest that modulating resistance-associated pathways, such as the AMPK/PD-L1 axis, may help overcome sunitinib resistance and enhance immunotherapy responses. Zhou et al. 49 investigated acquired sunitinib resistance in a U87MG glioma xenograft model and found that, although sunitinib suppressed angiogenesis-related gene expression, no significant difference in tumor vascular density was observed between sensitive and resistant groups. Instead, key pro-survival signaling pathways were aberrantly activated in resistant cells, with phospholipase C gamma 1 (PLCγ1) phosphorylation increased by 2.6-fold. These findings suggest that activation of alternative survival pathways contributes to tumor cell escape from sunitinib. Luo et al. 50 demonstrated that N 6 -methyladenosine (m 6 A)-modified tripartite motif-containing protein 37 (TRIM37) is significantly upregulated in RCC tissues and cell lines and is closely associated with sunitinib resistance. Mechanistically, TRIM37 promotes the degradation of SMARCC2 via the ubiquitin-proteasome pathway, thereby activating the Wnt signaling pathway and further driving the development of the resistant phenotype. 50 Mizumoto et al. 51 reported that in RCC cells, sunitinib can activate the epidermal growth factor receptor (EGFR), inducing epithelial-mesenchymal transition (EMT) and thus promoting the development of resistance. This study highlights the EGFR-mediated EMT pathway as a key mechanism underlying acquired sunitinib resistance in RCC. 51 Yao et al. 52 found that sunitinib downregulates the transcription factor GATA binding protein 1 (GATA1) in ccRCC, reducing its binding to the MIR885 promoter and suppressing miR-885-5p expression. This leads to derepression of perilipin 3 (PLIN3), resulting in increased lipid droplet accumulation and reduced tumor cell sensitivity to sunitinib. These findings highlight the complex and multifaceted mechanisms of sunitinib resistance in cancer models, involving metabolic reprogramming, survival pathway activation, and epigenetic regulation. While these mechanisms were identified in oncological contexts, they may also be relevant to their potential use in T2D. For instance, the activation of AMPK and modulation of lipid metabolism—both of which are involved in sunitinib resistance—also play central roles in T2D pathophysiology. Therefore, understanding and potentially overcoming these resistance mechanisms may not only improve sunitinib’s efficacy in cancer but also enhance its therapeutic potential in metabolic diseases.
Our MR analysis identified several novel causal genes associated with T2D, including PPARGC1A, ADIPOQ, IL-15, BDNF, and CHRDL1, each playing distinct yet interconnected roles in metabolic regulation, inflammation, and neuroprotection. These findings provide mechanistic insights into the pathogenesis of T2D and highlight potential therapeutic targets. PPARGC1A (Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1 Alpha) is a master regulator of mitochondrial biogenesis and energy metabolism. Reduced PPARGC1A expression in pancreatic islets has been linked to impaired insulin secretion in T2D patients.53,54 Genetic variants in PPARGC1A, such as the Gly482Ser polymorphism and promoter methylation, have been associated with altered insulin sensitivity and glucose homeostasis, suggesting both genetic and epigenetic mechanisms contribute to T2D pathogenesis.54–56 Our MR analysis further supports PPARGC1A as a causal gene for T2D, reinforcing its therapeutic relevance. ADIPOQ (Adiponectin) is a key adipokine with anti-inflammatory and insulin-sensitizing properties. Low adiponectin levels are strongly associated with T2D risk, while elevated levels correlate with improved metabolic outcomes. 57 Multiple SNPs in ADIPOQ have been implicated in adiponectin regulation and T2D susceptibility, highlighting its role as both a biomarker and a potential therapeutic target.58,59 Unlike previous observational studies that reported associations between ADIPOQ levels and T2D risk, our MR analysis provides evidence for a causal relationship, reinforcing ADIPOQ as a promising therapeutic target. Interleukin 15 (IL-15) is a pro-inflammatory cytokine involved in immune activation and T-cell function. Emerging evidence suggests that IL-15 signaling contributes to the development of autoimmune and metabolic disorders, including T2D. Studies have shown that blocking IL-15 signaling can reverse diabetes onset, suggesting that targeting this pathway may offer therapeutic benefits.60–62 BDNF plays a critical role in neuronal survival and synaptic plasticity and has been implicated in neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease. Recent studies have also linked BDNF to metabolic regulation, with reduced plasma levels observed in T2D patients and associated with poor glycemic control.63–65 Its role in mood regulation further underscores its potential as a target for managing diabetes-related mental health complications. Given its involvement in both neurodegeneration and metabolic dysfunction, BDNF may represent a key node in the crosstalk between diabetes and cognitive decline, offering a potential link between metabolic and neurological comorbidities. While CHRDL1 has been primarily studied in the context of nervous system development and repair, its emerging role in metabolic regulation remains underexplored. Our MR analysis identifies CHRDL1 as a novel candidate gene for T2D, suggesting a potential role in diabetes-related neuropathy. This finding expands the current understanding of CHRDL1 beyond its classical functions and highlights its relevance in metabolic disease. While SCARB2 and TPST2 were also identified as potential targets, their specific roles in T2D remain less characterized.66–70 Further functional studies are needed to elucidate their contributions to disease pathogenesis. Collectively, these findings highlight the complex interplay between metabolic regulation, inflammation, and neuroprotection in T2D and provide a foundation for future studies aimed at translating these insights into novel therapeutic strategies.
This study has several notable strengths. First, it represents the largest proteome-wide investigation to date, encompassing the analysis of 4907 plasma proteins, thereby providing a comprehensive and rich data resource for identifying novel T2D-related biomarkers and potential therapeutic targets. Second, the study includes a large number of T2D cases and has been independently validated in two geographically and ethnically distinct cohorts from Europe and East Asia, enhancing the generalizability and robustness of the findings. Third, by integrating multi-omics approaches—including proteomics, genomics, scRNA-seq, pathway enrichment analysis, and drug-target interaction assessments—the study offers a multidimensional perspective on the potential pathogenic mechanisms of candidate proteins in T2D, significantly advancing our understanding of the disease’s molecular underpinnings. Finally, external validation of the identified targets further supports their reproducibility and biological relevance, laying a solid foundation for future clinical translation and the development of targeted therapies for T2D.
Our study has several limitations that should be acknowledged. First, as with many large-scale omics studies, we primarily relied on summary-level proteomic and genetic data from the deCODE Genetics and FinnGen cohorts. While these datasets offer substantial statistical power and high-quality genetic instruments, they may obscure tissue- or context-specific effects of the identified proteins. To address this, we have incorporated scRNA-seq data to validate the expression of key hub genes in disease-relevant cell types, such as pancreatic beta cells and stellate cells. However, this approach does not capture dynamic regulatory processes or post-translational modifications, which are essential for understanding protein function and therapeutic potential. Further integration of multi-tissue omics and functional assays will be needed to fully dissect the context-specific roles of these proteins. Second, although we applied rigorous MR methods to infer causality, these analyses assume no horizontal pleiotropy. While we used stringent quality filters—such as instrument strength (R2 > 1%), outlier detection (MR-Egger, weighted median), and a recently developed method (BWMR) that accounts for pleiotropy by weighting SNPs based on their validity—we acknowledge that residual confounding cannot be entirely ruled out. Our findings should therefore be considered hypothesis-generating and require further experimental validation. In addition, the absence of in vitro and in vivo functional studies limits our ability to confirm the biological mechanisms or therapeutic efficacy of the identified targets. Future work should include experimental validation to establish causal pathways and evaluate the functional relevance of these proteins in T2D pathophysiology. Third, the majority of the datasets used in this study were derived from individuals of European and Asian ancestry. While we identified several promising candidates, the generalizability of our findings to other ethnic populations remains uncertain. Expanding our analyses to more diverse populations will be essential to ensure equitable applicability of the identified targets. Fourth, detailed demographic information—such as age, sex, and clinical subtypes—was limited in the available datasets. This restricts our ability to perform stratified analyses and assess how these variables may influence the observed associations. Future studies with access to more granular phenotypic data will enable more precise and personalized insights. Finally, while our multi-omics approach has uncovered novel therapeutic candidates, translating these findings into clinical applications will require extensive preclinical studies and well-designed clinical trials to evaluate the selectivity, specificity, and safety of targeting these proteins in T2D patients. In particular, we identified sunitinib as a potentially repurposable drug based on its known targets—such as VEGFR, KIT, and MET—which were among the proteins implicated in our MR analysis. However, we fully acknowledge that sunitinib is currently an FDA-approved tyrosine kinase inhibitor primarily used in oncology, and its safety and efficacy in treating metabolic disorders such as diabetes have not been clinically evaluated. We do not imply direct clinical applicability at this stage, but rather emphasize that preclinical studies—including in vitro systems, animal models of diabetes, and detailed toxicity assessments—are necessary before considering any translational applications.
Conclusion
In summary, our study identified numerous plasma proteins with strong causal relationships to T2D, providing new insights into T2D etiology and promising targets for biomarker screening and therapeutic drug development. Our findings suggest that sunitinib is a promising candidate for T2D treatment, and we validated the localization of hub genes using scRNA-seq data. Future research should prioritize validating these potential targets in vitro and in vivo, as well as conducting clinical trials to assess the efficacy and safety of sunitinib in T2D management. In addition, further exploration of the molecular mechanisms underlying these targets could lead to optimized therapeutic strategies and improved patient outcomes.
Supplemental Material
sj-pdf-2-tae-10.1177_20420188251376325 – Supplemental material for Proteome-wide and network pharmacology integration identifies sunitinib as a potential therapeutic for type 2 diabetes targets
Supplemental material, sj-pdf-2-tae-10.1177_20420188251376325 for Proteome-wide and network pharmacology integration identifies sunitinib as a potential therapeutic for type 2 diabetes targets by Yuan Kong, Hai-Wei Zhu, Hui-Xin Tong, Liang Shi and Hao Yu in Therapeutic Advances in Endocrinology and Metabolism
Supplemental Material
sj-xlsx-1-tae-10.1177_20420188251376325 – Supplemental material for Proteome-wide and network pharmacology integration identifies sunitinib as a potential therapeutic for type 2 diabetes targets
Supplemental material, sj-xlsx-1-tae-10.1177_20420188251376325 for Proteome-wide and network pharmacology integration identifies sunitinib as a potential therapeutic for type 2 diabetes targets by Yuan Kong, Hai-Wei Zhu, Hui-Xin Tong, Liang Shi and Hao Yu in Therapeutic Advances in Endocrinology and Metabolism
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.