
Abstract
Nonalcoholic fatty liver disease (NAFLD), also called metabolic dysfunction-associated steatotic liver disease (MASLD) is a prevalent syndrome marked by liver fat accumulation in the absence of significant alcohol consumption, encompassing simple fatty liver, nonalcoholic steatohepatitis (NASH), and advanced stages such as fibrosis and cirrhosis. Its incidence has surged globally, impacting up to 40% of the population, with a doubling of cases in China over a decade. NASH, a severe form, can progress to liver cirrhosis and cancer, posing a substantial health burden, especially among individuals with type 2 diabetes. Projections indicate a steep rise in NASH cases, necessitating urgent interventions beyond lifestyle modifications, such as innovative pharmaceuticals. Early diagnosis is crucial, yet current tools have limitations, highlighting the need for noninvasive, scalable diagnostic approaches. Advances in imaging and biomarker identification offer hope for early detection. Epigenetic factors play a significant role in MASLD pathogenesis, regulating key molecular mechanisms. Addressing MASLD requires a multifaceted approach, integrating lifestyle interventions, pharmacotherapy, and emerging therapeutics, against the backdrop of an evolving landscape in disease management.
Keywords
Introduction
Nonalcoholic fatty liver disease (NAFLD) refers to a syndrome characterized by liver parenchymal cell steatosis and fat accumulation without significant alcohol consumption. It includes simple fatty liver, nonalcoholic steatohepatitis (NASH), fatty liver fibrosis, and cirrhosis. In recent years, with the improvement in living standards, changes in lifestyle, and dietary structure, the incidence of NAFLD has been increasing year by year. NAFLD stands as one of the prevailing chronic liver conditions, impacting around a quarter of the global adult population. 1 Epidemiological studies show that NAFLD has become one of the most common chronic liver diseases globally, affecting 20%–40% of the human population. 2 In China, the number of affected individuals has nearly doubled compared to 10 years ago, especially in developed cities such as Beijing, Shanghai, and Guangzhou, where the prevalence is as high as 10%–25%, approaching that of some developed countries.3,4 Recently, a group of international experts issued a statement recommending that NAFLD could be renamed as metabolic dysfunction-associated fatty liver disease (MAFLD) in 2020. 5 Three years later, the term metabolic dysfunction-associated steatotic liver disease (MASLD) has been proposed. 6
NASH denotes the occurrence of liver fat degeneration concurrent with hepatocellular injury and inflammation, potentially progressing to liver cirrhosis and hepatocellular carcinoma, constituting a significant contributor to end-stage liver disease and liver transplantation. 7 Studies indicate that 20% of MASLD cases may progress to cirrhosis, with 30%–40% of patients dying from liver-related diseases; some patients may develop subacute liver failure and liver cancer. 8 In Japan, MASLD-related liver ailments, including cirrhosis and hepatocellular carcinoma, have emerged as the third leading cause of mortality among individuals with type 2 diabetes. 9 Projections suggest that by 2027, the populace affected by NASH in the United States, Japan, and five European Union nations (the United Kingdom, France, Germany, Italy, and Spain) will escalate to 18 million. 10 Presently, predominant approaches for managing NASH and MASLD revolve around lifestyle modifications, encompassing interventions such as dietary caloric restriction and physical activity. Nonetheless, sustaining these modifications proves arduous, underscoring the critical imperative for the development of innovative pharmaceuticals for the treatment of these diseases.
MASLD has become a ubiquitous health challenge, affecting a significant portion of the global population. Its insidious nature, often asymptomatic in the early stages, contributes to delayed diagnoses and increased disease burden. The escalating prevalence of MASLD amplifies its economic and healthcare impact, necessitating urgent attention to mitigate its consequences. Accurate and early diagnosis is pivotal for effective MASLD management. 11 Currently available diagnostic tools present limitations, and there is a pressing need for noninvasive, cost-effective, and scalable approaches. Advances in diagnostic technologies, such as imaging modalities and biomarker identification, hold promise for enhancing early detection and risk stratification. 12 The urgency to address MASLD extends to the development of interventions that span lifestyle modifications, pharmacotherapy, and emerging therapeutic modalities.13,14 Lifestyle interventions, while foundational, face challenges in long-term adherence. Innovations in pharmacological approaches and exploration of novel therapeutic agents underscore the dynamic landscape of MASLD management. Sarcopenia, a severe condition marked by the progressive loss of skeletal muscle mass and strength, is frequently associated with various chronic diseases, including NAFLD. This comorbidity is observed not only in obese individuals but also in lean NAFLD patients, reflecting its broad clinical impact. Sarcopenia exacerbates NAFLD through shared mechanisms such as systemic inflammation, insulin resistance, and mitochondrial dysfunction. 15 Despite its critical consequences, including disability, poor quality of life, and increased mortality, research on sarcopenia remains limited due to the scarcity of suitable animal models and the lack of standardized diagnostic criteria. Current diagnostic approaches, which rely on imaging and biomarkers, require further refinement and consensus.
The growing prevalence of MASLD demands an urgent and comprehensive response. This review highlights the relationship between epigenetics and MASLD has gained increasing attention. Studies suggest that various aspects related to the pathogenesis of MASLD, including adipocyte synthesis and differentiation, fat metabolism and transport, insulin resistance, and inflammatory factors, are extensively regulated by epigenetics. The key molecular mechanisms of regulation mainly involve DNA methylation, histone modification, and microRNAs (miRNAs).
This narrative review was conducted by systematically searching scientific literature from reputable databases, including PubMed, Scopus, and Web of Science, up to October 2024. The search strategy focused on key terms such as “Metabolic Associated Steatotic Liver Disease (MASLD),” “epigenetics,” “pathogenesis,” “NAFLD,” “sarcopenia,” and related topics. Priority was given to peer-reviewed articles published in high-impact journals, recent reviews, and seminal works in the field. The inclusion criteria emphasized studies that addressed the molecular mechanisms, clinical implications, and therapeutic strategies for MASLD. Articles were selected based on their relevance, methodological quality, and contribution to advancing the understanding of MASLD. Non-English articles and non-peer-reviewed materials were excluded to ensure scientific rigor. The current terminology comparison is shown in Table 1.
The current terminology comparison among NAFLD, MAFLD, and MASLD.
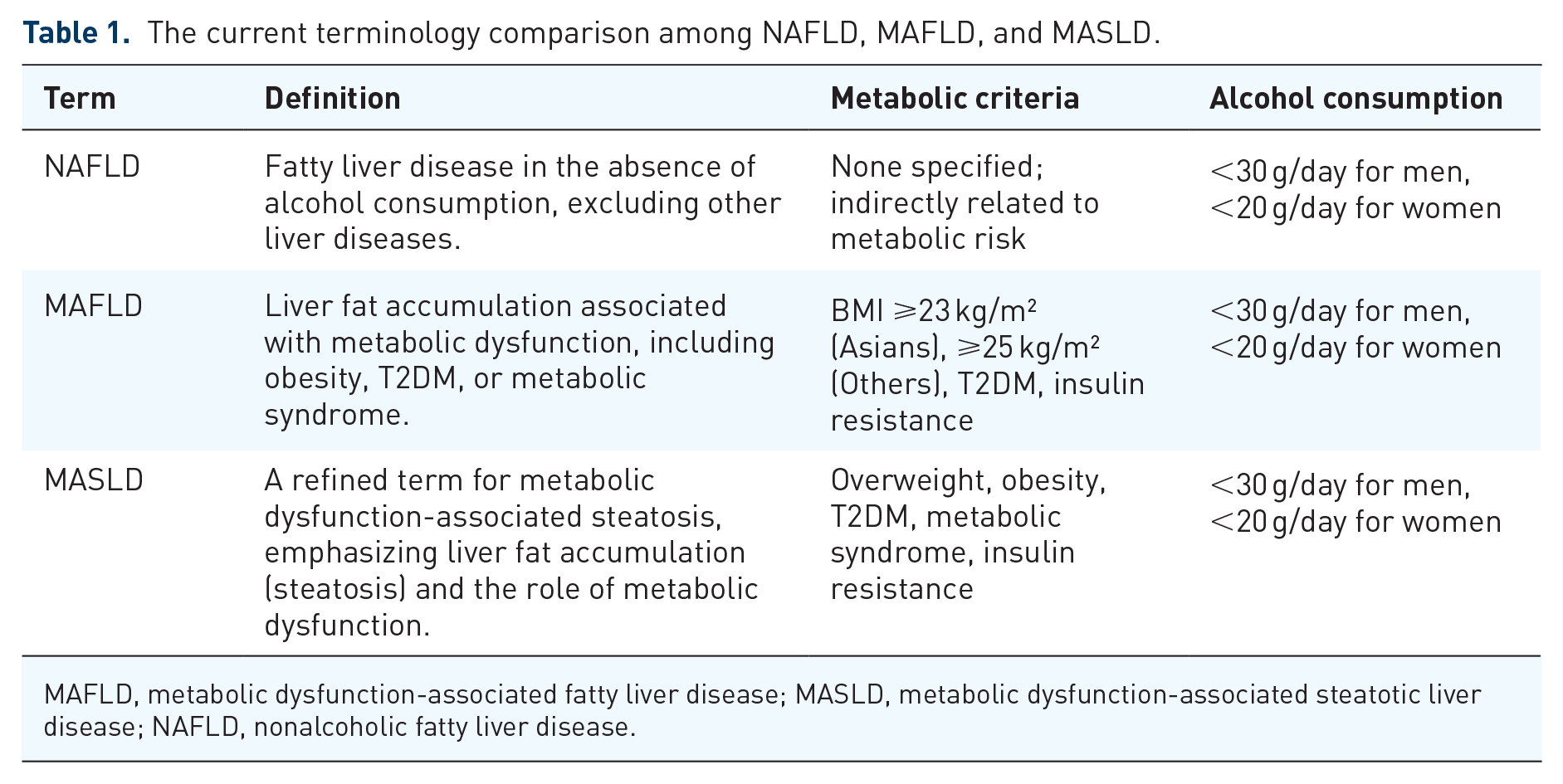
MAFLD, metabolic dysfunction-associated fatty liver disease; MASLD, metabolic dysfunction-associated steatotic liver disease; NAFLD, nonalcoholic fatty liver disease.
Understanding clinical and pathological characteristics
Clinical manifestations of MASLD
MASLD is a multifaceted condition with intricate pathogenic mechanisms involving lipid metabolism, insulin resistance, and inflammatory processes. A nuanced understanding of these pathways is crucial for identifying therapeutic targets and developing tailored interventions. Delving into the molecular and cellular intricacies of MASLD pathogenesis is paramount for effective and targeted therapeutic strategies. The clinical manifestations of MASLD can vary widely, ranging from asymptomatic to advanced liver-related complications.16,17 Here are some common clinical manifestations and the identification of subtypes with distinct clinical implications:
(1) Asymptomatic or mild symptoms: in the early stages, MASLD may be asymptomatic, and individuals may not experience noticeable symptoms. Some individuals with mild MASLD may have non-specific symptoms such as fatigue or discomfort in the upper right abdomen. 17
(2) NASH: NASH is a more severe form of MASLD characterized by inflammation and liver cell damage. Clinical features of NASH may include fatigue, abdominal pain, and an enlarged liver. NASH can progress to fibrosis and cirrhosis in some cases. 18
(3) Advanced fibrosis and cirrhosis: as MASLD progresses, it can lead to advanced fibrosis and cirrhosis. 19 Clinical signs of cirrhosis include jaundice, ascites, easy bruising, and muscle wasting. Individuals with cirrhosis are at an increased risk of liver failure and hepatocellular carcinoma.
MASLD can be classified into different subtypes based on the severity of liver damage and histological features. Subtypes may include simple steatosis (fatty liver without inflammation), NASH, and advanced fibrosis/cirrhosis (Table 2). Noninvasive methods such as imaging studies and serum biomarkers are used to identify and categorize subtypes. 12 Each subtype of MASLD has distinct clinical implications and requires tailored management strategies. 18 Individuals with NASH and advanced fibrosis/cirrhosis are at a higher risk of liver-related complications and may require closer monitoring and intervention. Identifying subtypes contributes to predicting disease progression and determining the appropriate level of medical interventions.
Diagnostic methods and prognosis of MASLD subtypes.

MASLD, metabolic dysfunction-associated steatotic liver disease; NASH, nonalcoholic steatohepatitis.
Pathogenesis of MASLD
The pathogenesis of MASLD is complex, involving a multifactorial interplay of metabolic, genetic, and environmental factors. At its core, MASLD represents a metabolic disorder closely tied to dysregulated lipid and glucose metabolism, which leads to ectopic fat accumulation in hepatocytes. In addition, the intricate cascade implicates inflammatory and profibrogenic macrophages in the advancement of liver fibrosis, potentially extending their influence on chronic inflammatory processes in various tissues. 20 This metabolic foundation is largely driven by insulin resistance, altered lipogenesis, and oxidative stress.18,21
(1) Insulin resistance as a central mechanism: Insulin resistance plays a pivotal role in MASLD development and progression. 18 Peripheral insulin resistance, particularly in adipose tissue, promotes lipolysis, leading to increased delivery of free fatty acids (FFAs) to the liver. This excessive influx of FFAs overwhelms hepatic β-oxidation and promotes their esterification into triglycerides, contributing to hepatic steatosis. Concurrently, insulin resistance in the liver exacerbates de novo lipogenesis (DNL) by upregulating transcription factors such as sterol regulatory element-binding protein 1c (SREBP-1c) and carbohydrate-responsive element-binding protein (ChREBP). The failure to suppress hepatic gluconeogenesis despite hyperinsulinemia further contributes to systemic metabolic dysregulation.
(2) Lipotoxicity and oxidative stress: accumulation of toxic lipid species, such as diacylglycerol and ceramides, initiates lipotoxic stress, leading to mitochondrial dysfunction and endoplasmic reticulum (ER) stress. 19 The resulting oxidative stress increases reactive oxygen species (ROS) production, damaging cellular structures and activating inflammatory pathways. ROS also promotes lipid peroxidation, generating reactive aldehydes that exacerbate hepatocellular injury.
(3) Adipose tissue dysfunction and inflammation: obesity-induced adipose tissue dysfunction is a critical contributor to MASLD. 21 Adipocyte hypertrophy and hypoxia in obesity lead to macrophage infiltration, creating a pro-inflammatory milieu. Adipose-derived cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), exacerbate hepatic inflammation and insulin resistance. Conversely, reduced levels of adiponectin, an anti-inflammatory and insulin-sensitizing adipokine, further impair hepatic lipid metabolism.
(4) The gut-liver axis and microbiome dysbiosis: Emerging evidence underscores the role of the gut-liver axis in MASLD pathogenesis.18,19 Dysbiosis of the gut microbiome alters intestinal barrier integrity, increasing the translocation of bacterial-derived products such as lipopolysaccharides into the portal circulation. These endotoxins activate toll-like receptor 4 on hepatic Kupffer cells, triggering inflammation and fibrosis. In addition, alterations in bile acid metabolism and short-chain fatty acid production influence hepatic lipid homeostasis.
(5) Genetic and epigenetic influences: genetic predisposition, particularly variants in genes like PNPLA3, 22 TM6SF2, and MBOAT7,23,24 contributes to interindividual variability in MASLD susceptibility and progression. 25 Epigenetic modifications, including DNA methylation and histone acetylation, influence gene expression in pathways related to lipid metabolism, inflammation, and fibrosis. For instance, hypomethylation of PPARγ promoter regions is associated with increased lipid accumulation in hepatocytes.
(6) Crosstalk between metabolic pathways: MASLD involves a dynamic interaction between hepatic and systemic metabolic pathways.26–28 Dysregulated glucose metabolism leads to hyperglycemia and compensatory hyperinsulinemia, fueling hepatic lipogenesis. Meanwhile, impaired fatty acid oxidation results in the accumulation of triglycerides and lipotoxic intermediates.29,30 The interplay between these pathways underscores the systemic nature of MASLD as a metabolic disorder.
Cutting-edge insights into epigenetic regulation in MASLD
The mechanisms of epigenetics intricately govern a wide array of physiological and pathological processes by orchestrating gene expression through adjustments in the accessibility of chromatin’s epigenetic code. 31 Crucially, epigenetic regulatory mechanisms are reversible, enabling lifestyle and environmental changes to dictate epigenetic patterns throughout life, providing dynamic modulation potential. Consequently, alterations in genes and proteins associated with epigenetics may represent promising avenues for prospective treatment strategies in clinical practice. 32
Epigenetic modifications can induce changes in the expression of genes related to lipid metabolism, inflammation, and oxidative stress, all contributing to the onset of MASLD. Molecular-level occurrences, such as cytosine and histone modifications and alterations in nucleosome localization, stand out as potential catalysts for epigenetic regulatory mechanisms. 33 An increasing body of evidence indicates that diverse epigenetic mechanisms significantly shape the progression of lean MASLD.19,34 Among these modifications, aberrant DNA methylation serves as an initiating event in the development of cancer in MASLD patients. 35 Moreover, the changes to the amino-terminal ends of histones play a pivotal role in preserving chromatin structure and governing gene expression in MASLD. 36 In addition, the evaluation of circulating miRNA profiles brings considerable promise as a noninvasive method for assessing and monitoring the severity of hepatic disorders. 37
DNA methylation and MASLD
DNA methylation is currently a well-explored regulatory mechanism in epigenetics. During the process of DNA methylation, cytosine protrudes from the DNA double helix and enters the gap at the binding site of cytosine methyltransferase. This enzyme transfers a methyl group from S-adenosylmethionine to the 5′ end of cytosine, forming 5-methylcytosine. 34 DNA methylation is a heritable gene modification that does not alter the DNA sequence. In higher organisms, DNA methylation typically occurs on cytosine residues within CpG islands, and the methylation of CpG islands can directly lead to the silencing of related genes. DNA methylation plays a certain role in the pathogenesis of MASLD.
At present, the precise mechanisms underlying the development and progression of MASLD are still not fully elucidated, but emerging evidence suggests that DNA methylation alterations may contribute to its pathogenesis. Numerous studies have identified specific DNA methylation patterns associated with MASLD. In a murine model, shifts in hepatic levels of DNMT1 and DNMT3A were recognized in association with the advancement of hepatic steatosis. 38 In addition, within human subjects, an increase in the expression of three DNA methyltransferases—specifically, DNMT1, DNMT3A, and DNMT3B—was noted in fibrotic livers. 39 In contrast, chronic liver disease exhibited a decrease in the hepatic expression of DNA demethylases, represented by TETs. These paired modifications led to noticeable fluctuations in the overall levels of 5-mC and 5-hmC, subsequently prompting widespread changes in transcription across the genome. 39 Oxysterols, known for their role as ligands activating receptors involved in lipid metabolism and inflammatory responses, emerge as notable contributors to the pathogenesis of MASLD. 40 The results demonstrated that the accrual of 25-hydroxycholesterol (25HC) within the nuclei of hepatocytes, cultivated under conditions of elevated glucose (HG), distinctly triggered the activation of DNMT1. This activation, in turn, resulted in a noteworthy elevation of 5-mC levels across approximately 2225 genes. These genes are intricately involved in 57 signaling pathways, encompassing pivotal processes such as those in the PI3K, cAMP, insulin, diabetes, and MASLD pathways. 41
Additionally, hypomethylation of the promoter regions of crucial lipid-regulating genes, such as peroxisome proliferator-activated receptor gamma (PPARγ), 42 has been observed in MASLD patients, leading to dysregulated lipid homeostasis. These molecules play pivotal roles in lipid metabolism and have been identified as central players in the pathogenesis of MASLD. The involvement of homocysteine in the epigenetic regulation of PPARα and PPARγ represents a critical aspect of MASLD pathogenesis. The interplay between homocysteine and PPARs suggests a multifaceted role in controlling hepatic lipid metabolism. It has been demonstrated that homocysteine induces DNA methylation, leading to the suppression of PPARα and PPARγ expression. This epigenetic modification, in turn, contributes to the dysregulation of adipocyte-specific genes. Building on this foundation, Ju et al. 43 extended our understanding by investigating the impact of demethylation specifically within the PPARγ gene. Their findings in obese rats exposed to a high-fat diet (HFD) revealed that demethylation of the PPARγ gene resulted in enhanced differentiation of adipocytes, providing mechanistic insights into the development of obesity. Previous research releases that betanin emerges as a potential therapeutic agent for steatohepatitis. 44 Its mechanism of action involves the upregulation of PPAR-α, downregulation of SREBP-1c, modification of adipokine levels, and modulation of the lipid profile. 44
This suggests a dynamic relationship between DNA methylation, PPARγ expression, and the differentiation of adipose tissue, shedding light on potential therapeutic targets for obesity-related conditions. Expanding the scope to the broader context of MASLD and its connection to metabolic syndrome, it becomes evident that DNA methylation plays a pivotal role in disease progression.
Furthermore, recent research indicates that mitochondrial dysfunction is involved in the progression of the pathogenic mechanisms of MASLD. 45 Recent study by Malik et al. 46 has shown that alterations in liver mitochondrial DNA (mtDNA) represent an early event preceding mitochondrial dysfunction and irreversible liver damage. Investigating the impact of HFD and Western diets (WD) on C57BL/6 mice for 16 weeks, the researchers observed diet-induced steatosis and an increased mtDNA content, particularly in the HFD group. However, the heightened mtDNA content in HFD mice was nonfunctional, evidenced by changes in mitochondrial proteins, while WD-fed mice exhibited accelerated liver dysfunction associated with alterations in mitochondrial oxidative phosphorylation and inflammatory pathways induced by mtDNA, suggesting a potential link between diet-induced mtDNA changes and the development of MASLD. 46
Furthermore, DNA methylation plays a crucial role in regulating the dynamic balance of key genes in glucose metabolism. Demethylation of the insulin gene promoter increases insulin production. Simultaneously, methylation of the insulin 2 promoter, when bound to methyl CpG-binding protein 2, leads to the silencing of the insulin gene, thereby inhibiting insulin production.47,48 This study of Monastero et al. 49 provides evidence of alterations in the methylation levels of the CpG island at the Vegfb promoter and Vegfb expression, both in vivo and in vitro, induced by dietary fatty acids, particularly linoleic fatty acid. The close correlation observed between Vegfb promoter methylation levels and Vegfb gene expression suggests that the regulation of Vegfb by dietary fatty acids may involve, at least in part, epigenetic modifications on the Vegfb promoter. Given the profound link between angiogenesis and tissue growth, the nutria-epigenetic regulation of Vegfb could serve as a crucial target for controlling adipose tissue expansion. 49 Therefore, alterations in DNA methylation can influence the expression of genes related to lipid metabolism pathways, playing a role in factors such as HFD-induced hepatic lipid deposition.
Moreover, DNA methylation alterations have been implicated in the progression from simple steatosis to more severe forms of MASLD, including NASH and fibrosis. 35 In a targeted DNA methylation study focusing on five genes involved in fibrogenesis modulation, the findings reveal hypomethylation of pro-fibrogenic genes (TGFβ1 and PDGFα) and hypermethylation of anti-fibrogenic genes (PPARα and PPARδ) in advanced MASLD compared to mild cases, suggesting that the methylation status of specific CpGs could potentially predict the severity and progression of MASLD to liver fibrosis. 50
Taken together, the advancements in our understanding of DNA methylation and its relation to MASLD have shed light on the complex molecular mechanisms underlying the development and progression of this prevalent liver disease (Table 3). Further research is warranted to elucidate the precise roles of DNA methylation in MASLD pathogenesis, identify novel biomarkers, and explore potential therapeutic interventions.
Summary of the key aspects of DNA methylation in MASLD pathogenesis.

25HC, 25-hydroxycholesterol; DNMT, DNA methyltransferase-1; HFD, high-fat diet; MASLD, metabolic dysfunction-associated steatotic liver disease; NA, Not Applicable; NASH, nonalcoholic steatohepatitis; PPARγ, peroxisome proliferator-activated receptor gamma; SREBP-1c, sterol regulatory element-binding protein 1c; TET, Ten-eleven translocation 1; WD, Western diet.
Advancements in understanding histone modifications and MASLD
Histone modifications, including methylation, acetylation, phosphorylation, and ubiquitination, are dynamic and reversible processes that regulate gene expression by modulating the structure and accessibility of chromatin. In the context of MASLD, aberrant histone modifications have been linked to the dysregulation of key metabolic pathways, inflammation, oxidative stress, and fibrogenesis.
Histone methylation has also been implicated in MASLD pathogenesis. Global changes in histone methylation patterns, particularly histone H3 lysine 4 (H3K4) trimethylation and H3K27 trimethylation, have been reported in MASLD. 36 These histone methylation alterations affect the expression of genes involved in lipid metabolism, inflammation, and fibrosis. Moreover, specific histone methyltransferases and demethylases have been identified as potential therapeutic targets for MASLD.
Early evidence has indicated that the H3K4 methyltransferase MLL2 may impact metabolism through random ENU mutagenesis. Studies have demonstrated that a germline Mll2 mutation in mice resulted in insulin resistance and impaired glucose tolerance. 51 In addition, in a different study, HFD feeding activated ABL1 kinase, leading to phosphorylation of PPARγ2 and increased interaction between MLL4 and PPARγ2. 52 This resulted in enhanced recruitment of MLL4 to the promoter of PPARγ2-regulated steatosis target genes, leading to increased H3K4 methylation and transcriptional activation, ultimately contributing to fatty liver development. 53 Moreover, histone methylation also regulates the activation of hepatic stellate cells (HSCs) involved in the transition from MASLD to NASH. In activated HSCs, the H3K4 methyltransferase ASH1 directly binds to the regulatory genomic regions of fibrogenic genes, facilitating H3K4 methylation and transcriptional activation. 54 Inhibiting ASH1 results in the downregulation of these fibrogenic gene expressions. 55
Histone acetylation stands out as an extensively investigated modification in the context of MASLD. Diminished levels of histone acetylation have been documented in diverse animal models and among individuals diagnosed with MASLD. This reduced acetylation correlates with the suppression of genes associated with fatty acid oxidation and insulin signaling, contributing to heightened lipid accumulation and insulin resistance within the liver. Conversely, experimental investigations have revealed that enhancing histone acetylation, particularly through the application of histone deacetylase (HDAC) inhibitors, holds the potential to alleviate liver steatosis and insulin resistance in MASLD. In HepG2 cells, stimulation by insulin led to a transient elevation in acetylation at the FASN gene promoter on H3 and H4, demonstrating cross-regulation with ChREBP, despite the unidentified histone acetyltransferase (HAT). 56 Moreover, the targeted reduction of NR2F6 in the liver mitigated hepatosteatosis and NASH in murine models by suppressing CD36 expression. 57 NR2F6 directly bound to the CD36 promoter, recruiting SRC-1, a constituent of the p300/CBP HAT complex, and facilitating H3 acetylation. Elevated expression of NR2F6 was noted in the livers of MASLD patients, a phenomenon subsequently diminished by metformin treatment in obese mice. Hence, the use of NR2F6 antagonists emerges as a prospective therapeutic strategy for MASLD by influencing histone acetylation. 57
The process of histone ubiquitination entails the specific attachment of ubiquitin molecules to histones, facilitated by enzymes during various ubiquitin-related processes such as activation, binding, and degradation. This modification exerts an impact on the conformation of chromatin, leading to the recruitment of downstream chromatin regulators. Noteworthy is the capacity of RNF20 overexpression to hinder pro-fibrotic factors, namely IL-6, TNFα, and VEGFA, thereby impeding TGF-β-induced hepatic fibrosis through the ubiquitination of H2BK120. 58 Furthermore, the phosphorylation of histone H3 serine 10 (H3S10) during mitosis, influenced by interactions between kinases and phosphatases, interfaces with other histone modifications. This phenomenon is exemplified in the ChREBP-mediated elevation of FASN expression, contributing to hepatic steatosis through histone acetylation, methylation, and H3S10 phosphorylation. 59 The paucity of extensive documentation on histone ubiquitination and phosphorylation in the context of MASLD underscores the need for further investigation. 59
Targeting histone modifications for the treatment of MASLD holds promise as a potential therapeutic strategy (Table 4). Numerous studies have investigated the use of small molecule inhibitors targeting histone-modifying enzymes, such as HDAC inhibitors and histone methyltransferase inhibitors, in preclinical models. These compounds have shown beneficial effects in reducing liver steatosis, inflammation, and fibrosis in MASLD. However, further research is needed to optimize the specificity and safety of these compounds for clinical use.
Histone modification in MASLD development.

ChREBP, histone acetyltransferase protein; H3K4, histone H3 lysine 4; H3S10, histone H3 serine 10; HAT, histone acetyltransferase; HDAC, histone deacetylase; IL-6, interleukin-6; MASLD, metabolic-associated steatotic liver disease; NASH, nonalcoholic steatohepatitis; PPARγ, peroxisome proliferator-activated receptor gamma; TNF-α, tumor necrosis factor-alpha.
MiRNAs and MASLD
MicroRNAs are a hotspot and challenge in the field of epigenetics, referring to a large class of small noncoding RNAs composed of 19–22 nucleotides widely present in eukaryotes. Their function involves binding to the 3′ untranslated region of target mRNAs, inhibiting or degrading their expression to exert biological functions. In the nucleus, miRNA genes are transcribed by RNA polymerase II to produce primary miRNAs, consisting of a 5′ cap, at least one hairpin structure of about 70 nucleotides, and a 3′ poly(A) tail. Subsequently, RNase III Drosha, assisted by the accessory factor DGCR8 (DiGeorge syndrome critical region gene 8), processes primary miRNAs into precursor miRNAs, removing sequences such as the 5′ cap, 3′ poly(A) tail, and flanking hairpin structures. The precursor miRNAs are transported from the nucleus to the cytoplasm by the action of transport protein 5. In the cytoplasm, the precursor miRNAs are further processed by another RNase III, Dicer, to generate short double-stranded RNA fragments, that is, mature miRNA chains and their complementary miRNA chains. Due to asymmetric thermal stability, the mature miRNA chain preferentially forms RNA-induced silencing complexes with other proteins, inducing silencing of endogenous mRNAs. The binding between miRNAs and target mRNAs in the seed region (positions 2–8) can result in complete or partial complementarity, leading to degradation or translation inhibition of the target mRNAs.60–62
It is speculated that there are as many as 1000 different miRNAs in the human body, regulating at least 30% of gene expression. Typically, a target gene can be coregulated by multiple miRNAs, and conversely, a single miRNA can regulate multiple target genes. 63 As a core component of gene regulatory networks, miRNAs extensively regulate complex processes such as adipocyte differentiation, lipid metabolism, and insulin resistance.64,65 Relevant miRNAs include miR-122, miR-103/107, miR-34a, miR-21, with miR-122 being the most extensively studied.
miR-122
miR-122 is a liver-specific miRNA, detectable as early as 12.5 days after fertilization, and its expression gradually increases postnatally, eventually reaching 70% of total liver miRNA. This indicates that miR-122 is the most important miRNA in regulating the liver.66,67 Elevated miR-122 levels were documented in instances of simple steatosis compared to healthy livers 68 and in cases of severe steatosis compared to mild steatosis. Conversely, diminished miR-122 levels were observed in the livers of individuals with both obesity and MASLD when compared to non-steatotic controls. 69 This reduction was evident in cases of NASH compared to “simple” steatosis 68 or healthy controls, as well as in situations of severe fibrosis compared to mild fibrosis. 70
Numerous studies have established a causal link between changes in miR-122 expression and the progression from NAFL to NASH. 69 Downregulation of miR-122 contributes to NASH-related fibrosis by influencing HSC activation pathways. 71 The long noncoding RNA NEAT1 is upregulated in HSCs during liver fibrosis, promoting HSC activation through the repression of miR-122 and subsequent de-repression of its target, pro-fibrotic transcription factor Krüppel-Like Factor 6 (KLF6). 71 This NEAT1/miR-122/KLF6 axis is implicated in human NASH based on analysis of cirrhosis liver biopsies.
MiR-122 expression is suppressed by TGF-β signaling in HSCs and fibrogenic fibroblasts. 72 Additionally, miR-122 exhibits an anti-fibrotic function by repressing the expression of fibrosis-related genes, including Fibronectin 1, alpha Smooth Muscle Actin, and Type 1 Collagen α1. The transcription factor CCAAT/Enhancer-Binding Protein α (C/EBPα) controls miR-122 expression in murine HSCs, and miR-122 negatively regulates collagen production.
Hepatic glutamine metabolism undergoes progressive de-regulation in NASH with increasing fibrosis severity. MiR-122 loss in mice enhances glutaminolysis, while miR-122 overexpression in hepatocytes blocks this pathway. 73 Glutaminolytic enzymes GLS and SLC1A5, upregulated in HSCs of fibrotic livers, are direct targets of miR-122.73,74 However, the role of miR-122 in regulating glutamine metabolism in HSCs/myofibroblasts and its impact on the fibrogenic phenotype remain to be determined.
MiR-122 is identified as a regulator of NASH-associated liver inflammation. MiR-122 knockout mice exhibit elevated production of pro-inflammatory cytokines and chemokines in hepatocytes, with RELB, a member of the NF-κB transcription factor family, identified as a direct miR-122 target. 75 Cholesterol loading of liver cells stimulates the release of exosomes containing miR-122, promoting an M1 pro-inflammatory phenotype in macrophages. 76 This suggests a potential link between miR-122, cholesterol, and NASH pathogenesis.
Novel miRNAs related to pathogenesis of MASLD
Recent studies elucidate the mechanistic involvement of novel hepatic miRNA species in the progression of MASLD, particularly focusing on miR-26a, miR-378, and miR-144.
(1) MiR-26a and ER stress: MiR-26a establishes a feedback loop with the ER stress response pathway orchestrated by Protein Kinase RNA-like Endoplasmic Reticulum Kinase (PERK). 77 In the context of healthy livers, the induced expression of miR-26a during ER stress mitigates the condition by specifically targeting eIF2α. Conversely, in individuals with MASLD, miR-26a experiences downregulation, exacerbating ER stress during prolonged metabolic challenges.
(2) MiR-378 and MASLD pathogenesis: implicated in both early and advanced stages of MASLD, miR-378 demonstrates upregulation in DIO mice and individuals with MASLD/NASH.78,79 LXRα effectively decouples miR-378 expression from PGC1β, resulting in the inhibition of mitochondrial function and fatty acid oxidation. Furthermore, miR-378 directly inhibits NRF1, leading to suppressed fatty acid oxidation and the promotion of hepatic steatosis. Notably, miR-378 also targets PRKAG2, contributing to the pathogenesis of NASH by fostering inflammatory signaling through NF-κB/TNFα.
(3) MiR-144 and hepatic lipid accumulation: In the context of obese and insulin-resistant individuals, miR-144 experiences upregulation in liver macrophages and hepatocytes.80,81 This upregulation results in the impairment of the antioxidant response to hepatic lipid accumulation by specifically targeting NRF2. Furthermore, miR-144 indirectly influences NRF2 activity by repressing IRG1 and modulating tricarboxylic acid cycle metabolites. Intriguingly, GATA4 is identified as a driving force behind the heightened transcription of miR-144 in the livers of obese insulin-resistant individuals, potentially induced by ERK signaling.
These miRNAs assume pivotal roles in the progression of MASLD, impacting ER stress, lipid metabolism, and inflammatory responses. A comprehensive understanding of their regulatory mechanisms offers valuable insights into potential therapeutic targets for the effective management of this widespread liver disorder.
Future perspectives
The understanding and management of MASLD continue to evolve rapidly. Future research should focus on unraveling the precise molecular mechanisms underlying MASLD progression, including its genetic and epigenetic drivers, to identify novel therapeutic targets. The development of reliable, noninvasive diagnostic tools, such as advanced imaging modalities and circulating biomarkers, is imperative for early disease detection and stratification of patients at higher risk of progression. In addition, the integration of artificial intelligence and machine learning in predictive modeling holds promise for personalized MASLD management. Therapeutically, emerging agents targeting key metabolic pathways, such as DNL inhibitors and gut microbiome modulators, warrant further exploration in clinical trials. Addressing comorbidities such as sarcopenia and cardiovascular disease through multidisciplinary approaches will also be critical for improving patient outcomes. Finally, global efforts are needed to harmonize nomenclature, diagnostic criteria, and treatment guidelines, ensuring equitable access to care and fostering collaboration in MASLD research and management.
Concluding remarks
In recent years, there has been growing evidence linking specific epigenetic changes to the pathogenesis of MASLD. One such epigenetic modification that has gained significant attention is DNA methylation. Studies have shown that aberrant DNA methylation patterns occur in genes involved in key metabolic pathways, including lipid metabolism, oxidative stress, and inflammation, which are all implicated in MASLD development. For instance, hypomethylation of the promoter region of the PPARγ gene has been associated with increased expression of this critical regulator of adipogenesis, leading to excessive lipid accumulation in hepatocytes. Histone modifications, another important epigenetic mechanism, have also been implicated in MASLD. Altered histone acetylation and methylation patterns have been observed in genes involved in lipogenesis, insulin signaling, and inflammation. These epigenetic changes affect the expression of key genes, disrupting the delicate balance of metabolic homeostasis and contributing to the development of MASLD. Understanding the specific epigenetic alterations associated with MASLD pathogenesis can provide critical insights into the underlying molecular mechanisms and potentially identify novel therapeutic targets for this complex disease.