
Abstract
Rett syndrome (RTT) is a neurodevelopmental disorder caused by mutations in the MECP2 gene, potentially disrupting lipid metabolism and leading to dyslipidemia (DLD) and steatotic liver disease (SLD). Although SLD has been described in RTT mouse models, it remains undocumented in humans. We herein describe a 24-year-old woman with RTT who was evaluated for abnormal liver enzymes. Imaging revealed hepatic steatosis, and transient elastography showed a controlled attenuation parameter of 342 dB/m and stiffness of 7.1 kPa. Laboratory investigations excluded secondary causes, including insulin resistance, metabolic syndrome, alcohol use, and new medications. Her Homeostatic Model Assessment for Insulin Resistance score was 1.8, her hemoglobin A1c concentration was 4.8%, and her lipid profile showed elevated triglycerides and low-density lipoprotein, consistent with DLD. Liver biopsy confirmed SLD. This case supports the hypothesis that MECP2 mutations in RTT disrupt lipid metabolism through a unique pathophysiologic mechanism, increasing the risk of DLD and SLD independently of traditional metabolic syndrome factors. It highlights the importance of early screening for liver disease in patients with RTT, despite their young age, to prevent complications. Additionally, it validates MECP2-null mouse models as reliable tools for investigating future therapeutic strategies in RTT.
Keywords
Introduction
Rett syndrome (RTT) is a rare and severe neurodevelopmental disorder that occurs almost exclusively in females, with an incidence of 1 in 10,000.1,2 It is primarily caused by loss-of-function mutations in the methyl CpG binding protein 2 (MECP2) gene, which maps to the Xq28 locus and encodes the MECP2 protein. 3 The mechanisms by which MECP2 variants cause RTT are not fully understood, but MECP2 deficiency is believed to result in abnormal synaptic maturation and maintenance in the brain. 1
Girls with RTT typically show what appears to be normal development during the first 6 to 18 months of life, followed by regression in developmental milestones. They progress through stages of stagnation and rapid regression during the early years, characterized by delayed and eventual loss of motor and language skills, hypotonia, stereotypic hand movements, microcephaly, seizures, and breathing irregularities. This is followed by a plateau period that can last for years or decades, eventually leading to significant motor deterioration. This final stage is marked by severe physical disability, dystonia, muscle wasting, scoliosis, and wheelchair dependence.4,5 However, the clinical presentation and symptom severity vary, likely influenced by factors such as non-random X-inactivation, mutation type, and modifier genes.1,5
The literature indicates that perturbed lipid metabolism in the nervous system is a key contributor to the pathophysiology of RTT, regardless of genetic background. This disruption affects critical neural tissue functions, including membrane trafficking, signal transduction, myelin formation, dendrite remodeling, neuropeptide formation, and synaptogenesis, ultimately driving the development and progression of neurological manifestations.1,2 However, as research into this syndrome advances, mutations in the MECP2 gene have also been found to contribute to the disease burden by causing disruptions in peripheral systems, leading to complications such as dyslipidemia (DLD) and steatotic liver disease (SLD).4,6,7 Although DLD has been described in human cell lines and individuals with RTT, SLD has only been reported in RTT mouse models.1,5,8,9
This report explores how peripheral lipid metabolism disruptions in RTT can present in clinical practice. We describe a patient with RTT who was evaluated for abnormal liver enzymes and subsequently diagnosed with SLD based on liver biopsy findings. The patient did not have insulin resistance or other comorbidities associated with metabolic syndrome, supporting the hypothesis that these disruptions result from a pathophysiologic mechanism specific to RTT. The CARE guidelines were followed during the preparation of this case report. 10 An abstract version of this case report was presented as a poster at the Canadian Liver Meeting. 11 Written informed consent for publication of this case report was obtained from the patient prior to submission.
Case report
A 24-year-old woman presented to a tertiary care hepatology clinic with a 6-month history of abnormal liver tests, showing a mixed pattern of enzyme elevation (alanine transaminase, 283 U/L; alkaline phosphatase, 258 U/L; and gamma-glutamyl transpeptidase, 1216 U/L) and a normal bilirubin level. Previously, regular blood work had revealed normal liver test results. Figure 1 illustrates the trend of the patient’s liver enzymes over time. Her medical history included genetically confirmed RTT with a T158M point mutation in MECP2, complicated by severe physical disabilities such as being non-verbal; being wheelchair-bound due to hypotonia, spasticity, and scoliosis; having neuromuscular respiratory failure requiring tracheostomy and ventilatory support; and having dysphagia managed with a gastrostomy tube for nutrition, seizures, and chronic pain. Additional diagnoses included protein-losing enteropathy, hypogammaglobulinemia requiring immune globulin replacement, central sleep apnea, hypothyroidism, osteoporosis, chronic iron deficiency anemia secondary to arteriovenous malformations, and duodenal strictures with prior partial small bowel obstructions. Abdominal ultrasound revealed moderately echogenic liver parenchyma consistent with hepatic steatosis, which was supported by transient elastography showing a controlled attenuation parameter of 342 dB/m and stiffness of 7.1 kPa. Investigations for secondary causes of liver disease, including hepatitis A/B/C serology, quantitative immunoglobulins, and autoantibodies, were unremarkable. The patient denied alcohol consumption, and drug-induced liver injury was deemed unlikely given the absence of new medications and no recent medication changes since 2019, aside from switching octreotide to a long-acting formulation. Her medications included stable doses of divalproex sodium since 2016, clobazam and modafinil since 2014, and Panax quinquefolius since 2018. Nutritional support consisted of Isosource Fibre 1.0 HP formula (775 kcal and 98 g protein per 24 hours) via gastrostomy tube, with no history of total parenteral nutrition. Regarding risk factors for SLD, her body mass index was 20.8 kg/m2 (weight, 48 kg; height, 1.52 m), her Homeostatic Model Assessment for Insulin Resistance score was 1.8, and her hemoglobin A1c level was 4.8%; additionally, her lipid profile showed elevated triglycerides (2.43 mmol/L) and low-density lipoprotein (LDL) (3.92 mmol/L), consistent with DLD. Figure 1 also shows her weight trend. There was no family history of liver disease, although both parents had type 2 diabetes and DLD. Consequently, a liver biopsy was performed.
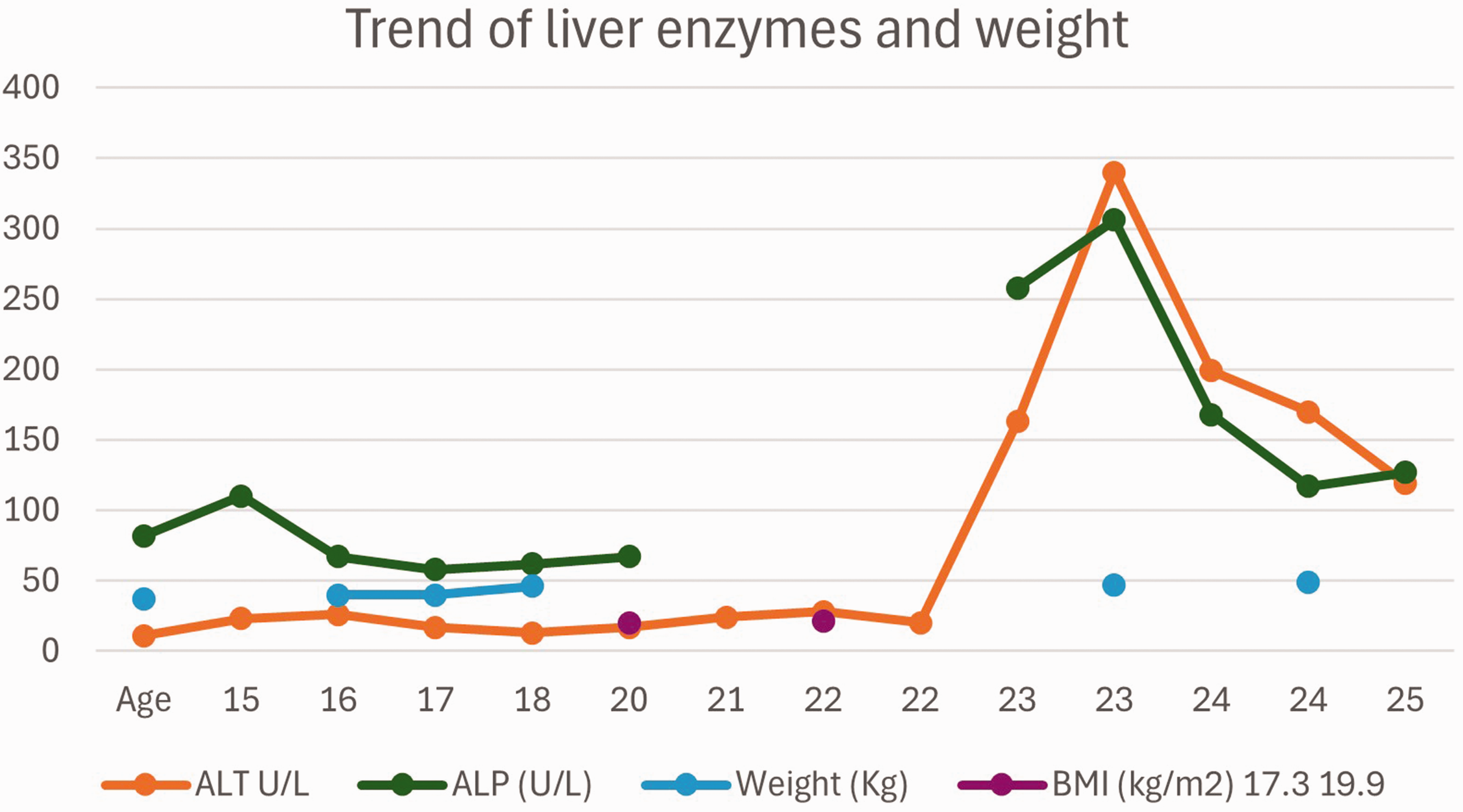
Trend of alanine transaminase (ALT), alkaline phosphatase (ALP), age, and body mass index (BMI) over time.
Liver biopsy (Figure 2) revealed moderate macrovesicular steatosis, focal lymphocytic lobular activity, and pericellular fibrosis, categorized as grade 1 and stage 1 according to the Brunt scoring system. There was no evidence of ballooning, interface hepatitis, ductopenia, or granulomas. The bile ducts appeared unremarkable, with no signs of cholestasis. Staining for copper, iron, and alpha-1-antitrypsin deposits was negative. Based on these findings, the diagnosis was confirmed as SLD, with the etiology attributed to RTT. In the absence of steatohepatitis or significant fibrosis, which are considered high-risk features for disease progression, treatment focused on dietary modifications. The patient provided consent for this approach.

Liver biopsy at 200× magnification, stained with hematoxylin and eosin. The lobules show moderate predominantly macrovesicular steatosis (approximately 60%), primarily in zone 2, with focal lobular activity and occasional lymphocyte aggregates. Portal tracts exhibit no significant inflammatory infiltrates, interface hepatitis, ductopenia, granulomas, Mallory-Denk bodies, ballooning, or cholestasis. Special stains reveal pericellular fibrosis (at most Stage 1) on Masson trichome, preserved cell plate architecture on reticulin, and no hyaline globules (negative RASD staining), iron deposits (negative Perls), or copper-associated protein deposits (negative rhodamine).
Discussion
This case describes a patient with RTT diagnosed with SLD on liver biopsy in the context of DLD, without insulin resistance or other comorbidities associated with metabolic syndrome. These findings suggest that her DLD and SLD may result from a pathophysiologic mechanism specific to RTT, potentially secondary to her MECP2 gene mutation causing disruptions in peripheral lipid metabolism.
Over the years, researchers have extensively used MECP2-null mice to model RTT and investigate the syndrome’s pathophysiology. The literature shows that lipid metabolism is significantly altered in both the nervous system and peripheral tissues when the MECP2 gene is partially or fully knocked out.1,2,6 Studies in hepatocytes demonstrate that MECP2 is essential for recruiting and anchoring the HDAC3-NCOR1-NCOR2/SMRT-TBLR1 complex to methylated DNA, thereby preventing transcription of target genes involved in lipid homeostasis.2,4,7 One key target gene is squalene epoxidase (SQLE), a rate-limiting enzyme in cholesterol biosynthesis. Failure to suppress transcription of SQLE and other lipogenesis enzymes leads to elevated serum levels of cholesterol, LDL, and/or triglycerides in MECP2-null mice compared with wildtype.12,13 Additional research shows that this dysregulation results in lipid accumulation in the liver and the development of SLD in these mouse models.1,2,6,14 Furthermore, studies indicate that these mice do not necessarily exhibit insulin resistance, further dissociating SLD development from metabolic syndrome and highlighting a distinct pathophysiologic mechanism in RTT. 6
Although limited data are available from human studies, some evidence suggests that patients with RTT also exhibit perturbed lipid metabolism, leading to elevated serum levels of LDL, cholesterol, and/or triglycerides.1,5,9 One study showed that these lipid abnormalities were not strongly associated with the body mass index and were more common in patients who had RTT with large MECP2 gene deletions, followed by the T158M missense mutation, indicating that this perturbation could be a metabolic manifestation of RTT. 1 Although the underlying mechanism is not fully understood, a study involving exome sequencing in two pairs of sisters with RTT identified a partial block in SQLE catabolism due to mutations in CYP24A1 and/or TM7SF2. These genes encode proteins that prevent SQLE accumulation and play an important role in lipid homeostasis. 8 This finding aligns with observations of increased serum lipid levels and the development of SLD in MECP2-null mouse models. However, no published reports to date confirm that this lipid metabolism disruption can lead to SLD in patients with RTT. Our case may be among the first real-world examples demonstrating this phenomenon in humans. Our patient, with a T158M mutation in the MECP2 gene, developed biopsy-proven SLD in the context of DLD as indicated by her lipid panel. This occurred without insulin resistance or other comorbidities associated with metabolic syndrome, further supporting the hypothesis that patients with RTT likely experience lipid metabolism disruptions similar to those observed in MECP2-null mice, increasing their risk of developing SLD. This type of steatosis may be better classified under the category of “specific etiology SLD” within the new umbrella term for SLD. 15 This case highlights the importance of screening patients with RTT for DLD and SLD at a young age to detect and address these conditions before complications arise. It also reinforces the validity of using MECP2-null mice as reliable models for identifying future therapies for RTT. 10 However, being the first reported case of its kind is a limitation because it remains unclear whether this finding represents a well-established association with RTT or a coincidental occurrence of SLD in a patient with RTT, given the high prevalence of SLD. Further investigation and increased awareness are needed to identify similar cases in other RTT patients and to clarify this association.
A common theme in the literature is that both neurologic and peripheral lipid metabolism are implicated in RTT.2,7,12 It has been hypothesized that serum findings in patients with RTT may correlate with processes occurring in the brain. 7 Consequently, serum lipid levels could potentially serve as non-invasive biomarkers to determine whether a patient might benefit from treatments targeting lipid metabolism.1,4,7–9 If so, interventions such as a ketogenic diet, lipid-lowering agents, and specific supplements could improve the metabolic, neurological, and behavioral phenotypes of RTT. Evidence supporting this has been demonstrated in mouse models. Studies have shown that lipid-lowering statin therapy reduces lipid accumulation, ameliorates motor symptoms, improves subjective health scores, and extends the lifespan of MECP2-null mice, with enhanced benefits when combined with adjuvant therapies such as ω-3 polyunsaturated fatty acids (PUFAs).1,2,4,7 Currently, there is no literature evaluating the role of statin therapy in patients with RTT. However, published case reports indicate that ketogenic dietary interventions can improve neurologic and behavioral phenotypes in patients with RTT. One study also revealed a significant reduction in the clinical severity of RTT and even partial rescue of symptoms in girls with stage 1 RTT after 6 months of ω-3 PUFA supplementation. 7
Conclusion
In summary, this case demonstrates that patients with RTT exhibit lipid metabolism perturbations similar to those observed in MECP2-null mouse models, leading to metabolic abnormalities such as DLD and SLD. The absence of insulin resistance and other comorbidities associated with metabolic syndrome further supports that these perturbations are caused by a pathophysiologic mechanism specific to RTT. If confirmed in other patients, these findings would advocate for screening patients with RTT for DLD and SLD. Additionally, our findings reinforce the use of MECP2-null mice as reliable models for identifying future therapies for RTT, including ketogenic diets, ω-3 PUFA supplementation, and lipid-lowering statin therapy.
Footnotes
Author contributions
LA: Conceptualization, investigation, writing (original draft), visualization. AA: Investigation, resources, writing (review and editing). AT: Investigation, resources, writing (review and editing). BH: Investigation, resources, writing (review and editing). VD: Investigation, resources, writing (review and editing). CMV: Conceptualization, investigation, writing (original draft), writing (review and editing), supervision, project administration.
Consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient.
Data availability statement
All data underlying the results are available as part of the article, and no additional source data are required.
Declaration of conflicting interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical considerations
Given the nature of the study, the Review Ethics Board of the University of Alberta waived the need of a full review.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.