
Abstract
This article aims to review current concepts in diagnosing and managing pheochromocytoma and paraganglioma (PPGL). Personalized genetic testing is vital, as 40–60% of tumors are linked to a known mutation. Tumor DNA should be sampled first. Next-generation sequencing is the best and most cost-effective choice and also helps with the expansion of current knowledge. Recent advancements have also led to the increased incorporation of regulatory RNA, metabolome markers, and the NETest in PPGL workup. PPGL presentation is highly volatile and nonspecific due to its multifactorial etiology. Symptoms mainly derive from catecholamine (CMN) excess or mass effect, primarily affecting the cardiovascular system. However, paroxysmal nature, hypertension, and the classic triad are no longer perceived as telltale signs. Identifying high-risk subjects and diagnosing patients at the correct time by using appropriate personalized methods are essential. Free plasma/urine catecholamine metabolites must be first-line examinations using liquid chromatography with tandem mass spectrometry as the gold standard analytical method. Reference intervals should be personalized according to demographics and comorbidity. The same applies to result interpretation. Threefold increase from the upper limit is highly suggestive of PPGL. Computed tomography (CT) is preferred for pheochromocytoma due to better cost-effectiveness and spatial resolution. Unenhanced attenuation of >10HU in non-contrast CT is indicative. The choice of extra-adrenal tumor imaging is based on location. Functional imaging with positron emission tomography/computed tomography and radionuclide administration improves diagnostic accuracy, especially in extra-adrenal/malignant or familial cases. Surgery is the mainstay treatment when feasible. Preoperative α-adrenergic blockade reduces surgical morbidity. Aggressive metastatic PPGL benefits from systemic chemotherapy, while milder cases can be managed with radionuclides. Short-term postoperative follow-up evaluates the adequacy of resection. Long-term follow-up assesses the risk of recurrence or metastasis. Asymptomatic carriers and their families can benefit from surveillance, with intervals depending on the specific gene mutation. Trials primarily focusing on targeted therapy and radionuclides are currently active. A multidisciplinary approach, correct timing, and personalization are key for successful PPGL management.
Keywords
Introduction
Pheochromocytoma and Paraganglioma (PPGL) is a rare neuroendocrine tumor that arises from chromaffin cells. Advancements in diagnosis have led to increased incidence from 0.19/100.000 per annum before 2000 to 0.58/100.000 per annum after 2010. 1
Recent progressions in clinical and laboratory medicine have allowed a much deeper understanding of PPGL, revealing new concepts and perspectives. Broad characterizations and rules, such as the ‘10% rule’ or the ‘5P disease’, have given way to precise genetic, molecular, biochemical, and imaging characterizations, making each PPGL a unique clinical entity.
The combination of very low incidence, complex clinical findings, and unpredictable tumor course makes PPGL a complex diagnostic and therapeutic challenge. 2 Surgical resection is still the treatment of choice. Timing of operation and pre-, peri-, and postoperative medical management remain topics of debate. Treatment of advanced disease and personalized approaches with targeted and radiotracer therapy are being thoroughly researched. 3
This review aims to present the most updated data on PPGL diagnosis and management.
Genetics
PPGL is considered the most heritable tumor since 30–35% of patients carry a germline mutation, and 40–45% have a somatic driver mutation in one of the more than 20 susceptibility genes.2,4,5 The genetic stroma influences tumor presentation, location, biochemical profile, and metastatic potential. Three common pathways, termed clusters, of tumorigenesis, have been identified, each leading to different characteristics summarized in Table 1.2,4,6–22 Additionally, there is ongoing research on several targeted therapies to address specific susceptibility genes, making personalized genetic testing even more important. 13 Meticulous case-by-case planning is crucial, as even the most established genotype–phenotype relationships are neither specific nor guaranteed.19,22–24 Different genotypes can have different phenotypic manifestations in various populations. 5 In addition, one gene mutation does not exclude the existence of another. In fact, gene combinations are not necessarily additive; they can be synergistic or permissive as well, resulting in a variety of phenotypes.7,15
Genetic and biochemical profile of PPGL with clinical associations.
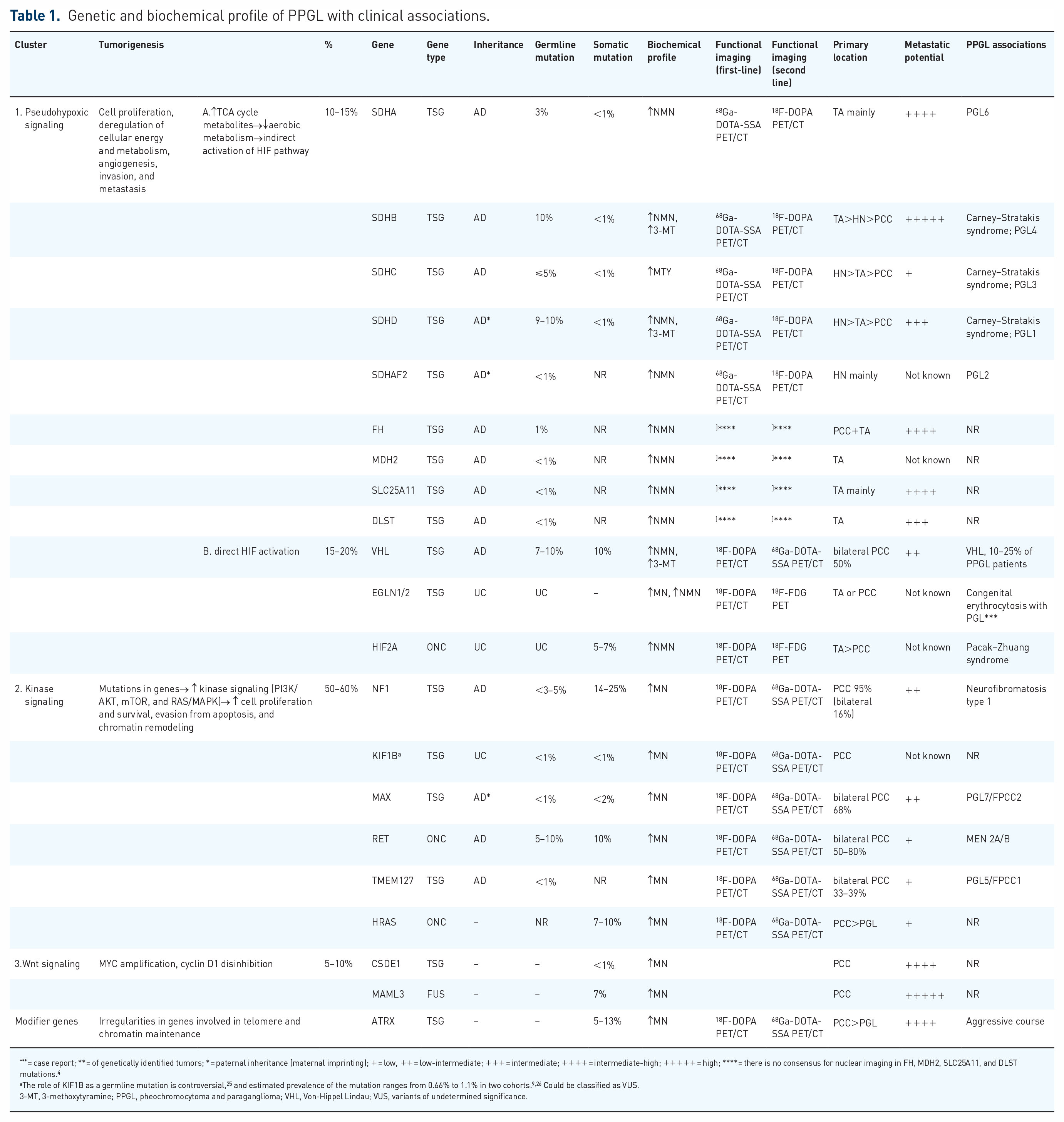
= case report; ** = of genetically identified tumors; * = paternal inheritance (maternal imprinting); + = low, ++ = low-intermediate; +++ = intermediate; ++++ = intermediate-high; +++++ = high; **** = there is no consensus for nuclear imaging in FH, MDH2, SLC25A11, and DLST mutations. 4
The role of KIF1B as a germline mutation is controversial, 25 and estimated prevalence of the mutation ranges from 0.66% to 1.1% in two cohorts.9,26 Could be classified as VUS.
3-MT, 3-methoxytyramine; PPGL, pheochromocytoma and paraganglioma; VHL, Von-Hippel Lindau; VUS, variants of undetermined significance.
Next-generation sequencing (NGS) advancements have led to its rise as a staple in genetic testing. Compared to sequential genetic testing (SGT), it allows physicians to test for specific gene panels based on each case simultaneously and cost-effectively. 27 Cost-minimization studies have linked NGS to fewer visits and up to 200 US $ in cost reduction relative to SGT. 28 The NGSnPPGL Study Group recommends the adoption of NGS as the new standard approach for PPGL genetic profiling. 29 These experts emphasize the need to develop universal standards for technical and interpretational aspects. Current limitations of NGS include the affordability of the most efficacious techniques, such as whole-exome and whole-gene sequencing, and the need for more consensus regarding variants of undetermined significance.30,31 NGS can also be beneficial for research purposes, assisting in discovering data that has yet to be interpreted. 27 This will lead to better incorporation of NGS in future PPGL management.4,29
Complete DNA profiling is vital for personalization and management guidance. Both tumor and blood (or saliva) samples are needed for a complete evaluation, as different germline and somatic mutations can coexist, and the distinction between them is of utmost importance. Furthermore, specific mutations can only be found on tumor samples and indicate metastatic potential (Table 1).23,24 With the exceptions of cases undergoing genetic evaluation secondary to family history or suspicion, guidelines suggest tumor sampling before blood (or saliva) sampling. Results can then guide the panel selection in serum samples, leading to a more efficient distinction between germline and somatic driver mutations. 30
With the current genetic landscape, 20–30% of tumors are still not associated with a known somatic or germline mutation. However, it is difficult to predict how many are ‘true negatives’ due to the lack of data. 4 Nevertheless, the absence of mutation does not exclude gene malfunction, and other mechanisms (e.g. abnormal splicing, promoter methylation) should be explored in patients where tumor characteristics follow a suspicious pattern. 32 If first-line testing fails, many promising markers related to gene expression are under evaluation for their clinical utility. These include various metabolome changes, as many PPGL mutations alter the Krebs cycle, and regulatory RNAs such as miRNA, lncRNA, and circRNA.33–36
Incorporating genetic testing as a standard part of PPGL management has led to better patient outcomes. A retrospective study by Buffet et al. 37 found that genetic testing improved follow-up rates, decreased the likelihood of severe metastases and metastatic potential, and resulted in better 5-year survival rates for patients with SDHx or Von-Hippel Lindau (VHL) mutations, particularly those with SDHB. The MAPP-Prono study, which involved 169 patients with metastatic SDHB-related PPGL, proved that active surveillance in this patient group attenuated the negative effect that was priorly attributed to this genotype. 38 Last but not least, the North American Neuroendocrine Tumor Society has deemed genetic testing an essential factor in determining the outcome of metastatic and unresectable PPGLs.39,40
Clinical presentation
The ‘modern era PPGL’ clinical presentation depends on a combination of interacting variables (Table 2). 4 In clinical practice, PPGL is discovered incidentally (incidentalomas) based on clinical features and in people with suspected or established germline mutations. 2
Factors determining the clinical presentation of diagnosis.

PPGL, pheochromocytoma and paraganglioma.
PPGL cases have been reported in newborns and individuals over 90 years of age, but most cases occur in the age range of 43–56.2 years.41–44 Cluster 1 mutation carriers tend to present symptoms earlier, typically between 20 and 30 years old (Table 1). 4 Incidentalomas occur later in life compared to clinically suspected PPGLs, and genetically suspected cases tend to appear even later.45,46
Recent research has challenged many traditional beliefs around PPGL. For instance, pulsatile features are not reliable indicators of PPGLs. 47 The frequency and intensity of symptoms can be influenced by various factors, both internal and external. Cluster 1 mutations are associated with consistent symptoms, while cluster 2 mutations often result in intermittent symptoms, also known as ‘spells’, corresponding to the nature of catecholamine secretion (Table 1). 4 Increased intrabdominal pressure, high-stress activities, and consumption of products rich in tyramine and drugs affecting CMN regulation can lead to paroxysms regardless of the tumor’s behavior. In addition, the classic triad of headaches, palpitations, and sweating is only present in 17% of patients, but each symptom on its own is more common. 48 That being said, the presence of the classic triad has the single greatest diagnostic value out of all PPGL manifestations. 49
PPGL incidence is higher in hypertensive patients than in normotensive patients (0.2–0.6%). 50 Inversely, hypertension (HTN) is present in up to 65–80% of PPGL cases.49,51 When diagnosing HTN, universal screening for PPGL is not recommended as it offers little insight. 49 PPGL should be suspected in unexplained, refractory cases or emergencies. Although BP positively correlates with CMN and plasma chromogranin A (pCGA) levels, tumor size does not significantly correlate with BP. 48 PPGL effects extend to the circadian rhythm of BP as well. Normal BP dipping, defined as >10% decrease between day and night measurements, can be present, absent, or reversed depending on various tumor characteristics such as malignancy or main CMN secreted.52,53 Last but not least, CMN surge can lead to HTN crisis with end-organ damage, particularly in undiagnosed patients. 54 While the different means of diagnosis do not have different prevalences, incidentalomas’ workup poses an additional risk factor as the dexamethasone suppression test can trigger HTN crisis in undiagnosed PPGL patients. Therefore, PPGL exclusion should precede testing of adrenal function. 55
PPGL can cause cardiovascular symptoms, including collapse, in up to 36% of patients. This can have the form of orthostatic hypotension, mainly due to CMN resistance 48 PPGL may induce Takotsubo cardiomyopathy syndrome (TS), which can cause left ventricular wall motion abnormality. Independent risk factors of PPGL-induced TS include chest pain, extra-adrenal tumor location, and female sex. Due to the diagnostic challenges and relative recency of the term TS, more concrete data are available for catecholamine-induced cardiomyopathy in pheochromocytoma and paraganglioma (CICMPP). Independent risk factors for CICMPP include tachycardia (⩾115 beats/min), SBP ⩾ 180 mm Hg, serum glucose ⩾8.0 mmol/L, ⩾3 PPGL clinical features, and age ⩽40 years. Dilated cardiomyopathy is the most frequent form of CICMPP (38.6%), followed by brady- and tachy-arrythmias, acute coronary syndrome, thromboembolic phenomena, myocarditis, and other forms of cardiomyopathy. 56
Hyperadrenergic effects can also lead to a hypermetabolic state with decreased visceral and subcutaneous fat and increased glycogenolysis. 51 CMN is also associated with anxiety, tiredness, constipation, diarrhea, nausea, flushing, tremor, and visual manifestations. ‘Sympathetic’ vasoconstrictive ischemic colitis has also been reported. Asymptomatic presentations are reported in up to 9% of patients. 48 A clinical score has been devised from all the above-mentioned symptoms that enables risk stratification based on clinical features. 46
Manifestations of extra-adrenal PPGL
Extra-adrenal PPGL features can be attributed to CMN excess or regional infiltrating phenomena and mass effect. 57 Parasympathetic and sympathetic chain PGLs are usually biochemically silent and rarely cause mass symptoms. 51 A classic location is the head and neck (HN) region.2,4 Up to 40% of them are hereditary; they are discovered incidentally in HN imaging or due to manifestations such as dysphagia, tinnitus, hoarseness, hearing loss, and other cranial nerve palsies. They most commonly infiltrate the carotid body and Cranial Nerve IX. 58 Besides the HN, PPGL can also arise in the bladder, presenting with lower urinary tract symptoms such as urgency, hematuria, and micturition ‘spells’. Bladder PPGLs need special attention from urologists, as they can trigger massive HTN crises during surgery if unsuspected. 59
Laboratory evaluation
Initial screening and confirmatory testing
Except for critically ill patients and patients who are suspected of having silent tumors, biochemical confirmation of PPGL is a prerequisite before any imaging investigation. 60
The most appropriate tests for initial screening and confirmation are plasma or 24-h urine free metanephrine/normetanephrine (MN/NMN) levels. Plasma 3-methoxytyramine (3-MT) levels can increase the diagnostic sensitivity (SE) up to 28% 61 in tumors suspected to secrete dopamine (DA) but are otherwise not included in testing. 62 Current guidelines suggest that both plasma free metanephrines (PMets) and urinary free metanephrines (UMets) are equally SE (97%) and SP (91%). 3 However, there are several indications of PMets being more SE but less SP than urinary free UMets, making them more valuable diagnostically for high-risk patients.51,63 Thus, if not measured simultaneously, PMets should be the initial step in management. 64
Ideal sampling
Proper sampling is vital in achieving maximal diagnostic accuracy. As in most cases, blood should be drawn in a fasting state. When 3-MT levels are co-tested, foods high in tyrosine should be avoided at least 3 days prior. 51 Furthermore, various drugs can alter CMN secretion and metabolism of CMN and DA (Figure 1). 65 Thanks to the advancement of laboratory techniques, however, patients can continue regiments including these drugs throughout testing, only considering temporary withdrawal on a case-by-case basis after the results. 3 Minimizing sympathetic activity is also essential for the diagnostic accuracy, particularly for PMets. Current guidelines recommend at least 20–30 min in the supine position before blood testing to further limit false positive (FP) results. Physical exercise should also be withheld before testing.3,66,67 If patients fail to meet the above requirements, they should only be evaluated through UMets testing. 2

Pharmacodynamic interferences increasing diffusion and metabolism of Epinephrine/NorEpinephrine and thus MN/NMN and 3-MT.
Laboratory advancements
Liquid chromatography with tandem mass spectrometry (LC-MS/MS) is the ‘gold standard’ compared to liquid chromatography with electrochemical detection (LC-ECD) and immunoassay (IA). LC-MS/MS has several advantages regarding clinical applications, lower consumable costs, and simplified sample preparation. 68 Analytic interference, although minimal, is mainly caused by ion suppression/enhancement, ion cross talk, and isobaric interferences. Maximizing sample purification and chromatographic resolution is critical in neutralizing analytical interference. 68 Finally, other drugs that can cause analytical interference are summarized in Table 3.
Analytical interferences of relevant drugs with LC-MS/MS.

Source: Table adapted from Davies and Davison. 68
3-MT, 3-methoxytyramine; LC-MS/MS, liquid chromatography with tandem mass spectrometry; MN, metanephrine; NMN, normetanephrine
Personalized reference intervals and interpretation
Minimizing inaccuracy substantially depends on reference interval (RI) personalization. Anthropometry and demographics should be taken into consideration. Age-specific RIs statistically significantly eliminate FP results of free plasma NMN. Gender-specific RIs are not required for plasma measurements, contrary to UMets, where they significantly improve SP. Plasma MN and 3-MT Upper Level - ULRIs should be set above the 99.5th percentile.2,69 Decreased glomerular filtration also affects both UMets and PMets, and UlRIs should be decreased and increased accordingly. 70 Even with appropriate adjustments, a few scenarios can still provide challenges in clinical practice.
While the Negative Predictive Value of both urinary and plasma measurements is nearly perfect, there are some scenarios that physicians should be aware of, such as silent or pseudo-silent PPGL (mainly Head & Neck Paraganglioma (HNPGL). 51 Diagnosis of these tumors should primarily rely on imaging and genetic suspicion. Imaging and further testing should be carried out in any case with negative biochemical testing and retained clinical suspicion. NETest, pCGA, and other neuroendocrine proteins can also be tested and are particularly useful for silent SDHB-related PPGL.61,62,71
The most challenging scenario is undoubtably borderline positive results (one–threefold ULRI). 68 These tumors need careful stepwise clinical, biochemical, and imaging approach. Adequate sampling must be ensured in all borderline patients and corrected if necessary. Pre-test prevalence is also a crucial determinant of interpretation, and the need for re-evaluation and follow-up. 3 Clinical suspicion is also a powerful guidance tool; low-suspicion patients should be followed up for at least 6 months, as sustained or elevated levels of free metabolites are highly suggestive of a tumor. Patients with moderate or high clinical probability may benefit from clonidine suppression test (CST).72,73 Finally, similar patterns in paired PMets and UMets in borderline cases indicate PPGL and should lead to further investigation. 72
Besides being significant indicators of PPGL, Pmets and UMets are also valuable in predicting tumor profiles. Tumor size has been positively correlated with MN and NMN levels (r = 0.81 and r = 0.71, respectively, p < 0.001), while higher MN/NMN ratios indicate adrenal tumors. 74 Conversely, elevated 3-MT levels suggest extra-adrenal disease and higher metastatic potential. 61 Figure 2 is a proposed algorithm that outlines a step-by-step approach to plasma and urine free metabolites.

Suggested step-by-step approach for plasma and 24-h urine free metabolites interpretation.
RIs should also account for the means of presentation in PPGL.46,48 Statistically significant differences between incidental, symptom-based, and mutation-based diagnosis for PMets, UMets, and isolated plasma free NMN levels are reported in multiple series.45,46,48 These differences should always be considered, particularly in borderline cases (Figure 2).
Additional testing
Even though CST is less incorporated in current clinical practice, guidelines suggest its use for controversial results in first-line testing.2,3,51 When used appropriately, Specificity and Positive Predictive Value can be as high as 100% in some prospective studies. 75 The efficacy of neuroendocrine markers such as Cromogranine A (CGA), Neuron Specific Enolase (NSE), and synaptophysin is controversial. The addition of pCGA measurement has mixed effects on SE and SP in different series.76,77 pCGA can be used as a last resort option for inconclusive diagnostic results. The same principles apply to NSE and synaptophysin, although rarely used. Patients undergoing plasma CGA measurements should discontinue protein pump inhibitors for 2 weeks to avoid FP results. 78
Recent evidence supports the utility of the NETest, a liquid mRNA biopsy used in NET diagnostics. The NETest uses a 51-gene multigenomic assay divided into 14 omic clusters representing tumorigenesis’ most crucial hallmarks. NETest’s uniqueness lies in its ability to quantify gene expression and thus provide an even more specific and personalized outlook on important tumor aspects, such as behavior and metastatic potential.79,80 While further prospective evidence comparing NETest to more significant clinical biomarkers is needed, recent evidence shows that NETest can detect PPGLs with an accuracy of up to 94% and provide valuable information about tumor characteristics. For instance, cluster 1 mutations have been associated with statistically significantly lower NETest scores than cluster 2 mutations. In addition, PHEOs have been associated with higher scores compared to PGLs. Metastatic tumors are also associated with higher scores than locoregional diseases. 81 Most importantly, the NETest relies on mRNA expression rather than secretory status, making it useful for biochemically silent masses and tumor follow-up. This principle’s benefit extends to clinical practice, as it requires minimal blood (1 mL) and is unaffected by patient position, lifestyle, and medications. 79 When compared to pCGA, NETest has been reported to be more accurate for diagnosis, staging, and predicting metastasis and recurrence. 81 The only potential downside of the NETest is that its overall sensitivity for NETs can lead to ‘false positive’ PPGL results when isolated. However, this has relatively low clinical significance, as the differential for PPGLs based on clinical appearance, imaging, and biochemistry is relatively narrow. Thus, many potential confounders can confidently be eliminated (see Future Directions).
Imaging
Anatomical imaging of adrenal tumors
Although CT and magnetic resonance imaging (MRI) can be used, CT is usually preferred as it is more cost beneficial and has better spatial resolution. 82 CT has a total sensitivity of 90% for adrenal gland tumors. PCC can be homogenous or heterogenous, with liquid-solid components and hypervascularity elements. Tumor size usually ranges from 1 to 15 cm, and smaller tumors tend to have homogenous profiles, while larger ones tend to be associated with central necrosis. High attenuation in CT is related to macroscopic fat accumulation, calcification (up to 7% of patients), or hemorrhage. PCC can be cystic and giant cystic as well. 83 While these findings can also be found in adrenal adenomas, a characteristic finding for PCC is unenhanced attenuation of >10 HU.84,85 Due to the polymorphic and unspecific nature of adrenal incidentalomas, contrast enhancement does not seem to be distinguishing but can be localizing in 88–100% of cases. PCCs tend to enhance more on the portal venous than the arterial phase. That being said, 110–120 HU enhancement in the arterial phase indicates PCC or hypervascular metastases. 86
Despite being the more sensitive option, MRI should not be the first-line option for PCC depiction. Nonetheless, it is more suitable for children, pregnancy, CKD, and contrast-medium allergy. It should also be considered in patients with metastatic disease, germline mutation, and recent excessive radiation exposure. Several findings have been associated with PCC but with limited diagnostic accuracy. Despite occurring in only one-third of the patients, T2-weighted high-intensity signal is relatively specific for PCC. 87 In cystic PCC with central necrosis, this high-intensity is sometimes referred to as a ‘light bulb’ lesion.
Anatomical imaging of extra-adrenal and metastatic tumors
CT and MRI are still the options for anatomical imaging of extra-adrenal and metastatic tumors. MRI appears to be more sensitive for detecting extra-adrenal PGLs, providing further details of the morphology of the tumor. 88 Thus, MRI is superior to CT for recurrent, residual, and metastatic tumors, except the lungs, whereas CT is more sensitive for primary pulmonary paraganglioma and pulmonary metastases. 89 HNPGLs need a combination of temporal bone CT, MRI, and MRA for adequate localization. MRA 3D time-of-flight and 4D MR acquisitions are highly sensitive for tumor detection. 90
Functional imaging
SE and SP improve substantially when anatomical imaging is combined with functional imaging. Planar scintigraphy, SPECT, and PET are the most frequently used options. PET is the best choice, as it has the shortest injection-to-imaging interval, scanning time (approximately 2 h), and the highest spatial resolution. 91 PCCs express various transporters on a cellular level, which correlates to multiple radiopharmaceutical components being able to give vast and diverse functional information about tumors (Figure 3). The choice of radionucleotide is personalized and depends on the tumor’s biochemical and genetic profile, location, and size (Table 1, Figure 4). 92

Radiotracer target receptors expressed on PPGL cells.
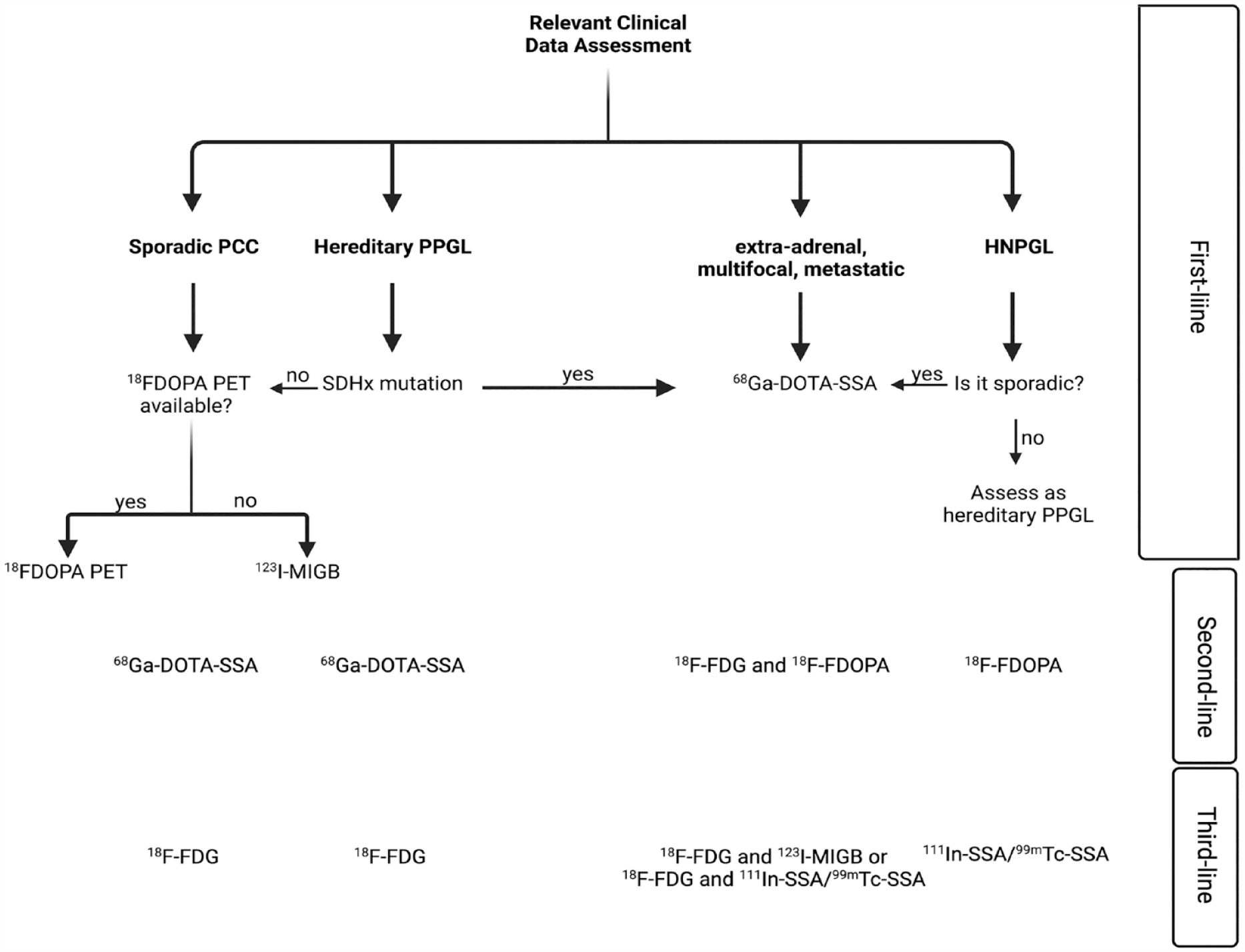
Suggested algorithm for functional imaging in PPGL.
123 I-MGB is very useful in addition to biochemical profiling and anatomical imaging as it is highly SE and SP for nonmalignant PCCs, especially when combined with SPECT. 91 It can locate tumors larger than 1 cm in diameter but cannot consistently detect smaller tumors or necrotic features. While 123 I-MGB sensitivity for adrenal tumors is 83–100%, it drops to 52–75% for PGLs and even lower, 18–50%, for HNPGLs. 51 Regarding metastatic disease, 123 I-MGB scintigraphy should be considered when peptide receptor radionuclide therapy (PRRT) with 123 I-MGB is a potential treatment option. 92
The increased rates of glycolysis allow the usage of 18 F-FDG PET. It can be used to visualize many tumors, especially malignant ones. Its uptake is independent of CMN secretion, allowing for silent tumor identification. 18 F-FDG PET is also useful for cluster 1A mutations that alter glucose metabolism and can substitute for all PPGL tumors (Table 1).92,93 Alternatively, 18 F-DOPA helps detect CMN-producing tumors. 18 F-DOPA is highly SE and SP for PCC and is currently the functional imaging of choice when available. Compared to 123 I-MGB, it allows for better clinical data through PET/CT. It is also recommended as first-line for the majority of hereditary tumors as well as for polycythemia-related PPGLs, FH, or MAX mutations (Table 1).93,94
SST analogs, primarily 68 Ga-DOTA-SSA PET, might be the most all-around out of all the options (Figure 4). It is the first-line option for non-adrenal and metastatic PGLs, HNPGLs, and multifocal disease. 92 68Ga-DOTA-SSA was found to have the highest detection rate (98.6%) out of all imaging options for patients with SDHB and generally all SDHx mutations (Table 1). As with 123 I-MIGB, SSA functional imaging is also indicated in potential candidates for SSA PRRT with 177Lu and 90 Y.95–97
Diagnosis
Identifying high-risk subjects
The low incidence of PPGL combined with the potentially fatal complications of a missed diagnosis and the costly workup make identifying high-risk subjects a top priority in management. According to current guidelines, PPGL investigation is recommended in patients with characteristic signs and symptoms, spontaneous or provoked, signs of cardiovascular instability suggestive of PPGL, and new-onset T2DM in lean subjects (BMI < 25 kg/m2). Density ⩾ 10 HU on CT imaging of an adrenal incidentaloma is also a high-risk feature. Established carriers of a germline mutation in one of the susceptibility genes, personal or family history of PPGL, and syndromic features suggesting PPGL should also be considered when screening candidates. 2
Changing perspective
Advancements in diagnostic testing, the goal of early identification in the asymptomatic phase, and the order in which the diagnostic pillars are put together have changed the landscape of PPGL. Moreover, moving away from the ‘10% or 5P disease’ to an incidental finding in imaging combined with the advancements in genetics have altered the context of PPGL throughout the last century. More than 80% of PPGLs until the 1970s were diagnosed due to symptoms, particularly unexplained HTN or features of the classic triad. Several studies report that over 60% of PPGLs are due to incidental discovery (Figure 5).1,45,46,48,98

A paradigm shift in the method of diagnosis for PPGL over the last decades, with the associated exponential trendlines.
Another massive shift in the field of PPGL diagnosis is personalization, following the general trend of oncology. Future research should be directed toward establishing guidelines for physicians, where specific tumor characteristics will be required to make a diagnosis. We propose a diagnostic algorithm/checklist based on the current research and future goals (Figure 6).

Proposed personalized diagnostic algorithm for PPGL.
Metastatic disease
Metastatic or malignant PPGL represents 10–20% of patients with these tumors. 2 About 10–15% are present at initial diagnosis. 99 Approximately 50–70% are sporadic, while others are due to susceptibility gene mutations, particularly SDHB (Table 1). More than 90% of malignant tumors are functional, with NA or DA biochemical profiles.4,10,38,100
Clinical manifestations are not different from benign PPGL. 4 Nonetheless, tumor burden can cause more significant CMN fluctuations leading to more ‘spells’ and cardiovascular collapse incidents. 101 Risk factors for metastatic disease are genetic profiles (Table 1), tumor size > 5 cm, multifocal disease, and NA or DA biochemical profile highly correlated with the genetic profiles and personal history of metastasis or recurrent PPGL.2–4,51 These factors and age at diagnosis also predict outcome and survival. 38
Currently, the best imaging approach for metastatic PPGL detection is 68 Ga-DOTA-SSA PET, with 18 F-FDG and 18 F-FDOPA being the second and third lines, respectively (Figure 4). Anatomical imaging is optional unless surgery is feasible. 2
Treatment and management
Localized disease
PCC surgery
When feasible, adrenalectomy is the standard of care for PCC patients, as it is the only potentially curative treatment.2,4,51,102 Surgical exploration of PCC has shifted massively from open adrenalectomy (OA) with an anterior transperitoneal or a posterior retroperitoneal incision to minimal invasive surgery (MIS) using either transabdominal or retroperitoneal approaches.3,103 The retroperitoneal approach is especially favorable for simultaneous bilateral adrenalectomy. 104
Current clinical guidelines state that laparoscopic approaches must be considered the standard of care, although no evidence comes from randomized controlled trials (RCT). Retrospective series suggest that laparoscopic adrenalectomy (LA) is a shorter procedure than OA, albeit not statistically significantly. 105 However, LA is associated with a statistically significant reduction in estimated blood loss (EBL).105,106 MIS has resulted in shorter hospital stays, better pain tolerance, faster recovery periods, and reduced perioperative morbidity. The complication rate is significantly lower as well. Most importantly, this favorable surgical profile is also associated with comparable oncologic results to OA, even for tumors with large diameters.106,107
With minimal invasive adrenalectomy prevailing as the better option, it is crucial to establish the efficacy of robotic adrenalectomy (RA) compared to LA. A 2022 meta-analysis, including 2985 patients with any indication for adrenalectomy suggests that RA is statistically superior to LA in EBL (WMD = −18.25, 95% CI −27.85, −8.65, p < 0.01), length of stay (LOS) (WMD = −0.45, 95% CI [−0.57, −0.33], p < 0.01), and conversion to open (OR = 0.31, 95% CI 0.12, 0.78, p = 0.01). However, only reduced LOS (WMD = −0.49, 95% CI [−0.83, −0.15], p < 0.01) was reported in patients with PCC. Since both methods produce similar results, cost and expertise should be the determining factors of choice. Cost-benefit analysis literature comparing RA to LA is limited, and a recent retrospective study showed that the calculated relative costs fees were not increased in RA compared to LA for an experienced surgical team. 108 However, results can contradict.109,110
Advances in diagnostics have shown that bilateral disease is more often than previously assumed. Thus, partial adrenalectomy (PA) should always be considered in the case of familial PCC due to the minimal likelihood of ipsilateral recurrence and avoidance of chronic glucocorticoid replacement. 111 Individualization is critical in weighing the pros and cons. Reoperation with a retroperitoneal approach is almost always feasible and independent of the first operation. 112 While more evidence is needed for safe conclusions on nonhereditary PCC, current guidelines suggest PA for low-risk hereditary PCC (VHL and RET) with small tumors and a history of complete contralateral adrenalectomy. 3
Surgery is the most critical factor in successful treatment. Complete resection is to be achieved without tumor rupture and en block. The tumor should not be heavily manipulated as this can trigger CMN surge and lead to cardiovascular collapse. Adrenal vein ligation should also be noted to the anesthesiology team, as prolonged ligation can lead to hypotension. The timing of adrenal vein ligation during the adrenalectomy seems irrelevant. 113
PGL surgery
The complete resection of HNPGLs primarily depends on anatomic characteristics and patient performance status. 4 It is generally reserved for young patients with no comorbidities, as these tumors are usually well tolerated by the elderly. Prior embolization of the tumor may be indicated. Depending on the length of the intradural space (IDS) or internal carotid artery (ICA) involvement, surgical excision may need to be carried out in multiple steps. 114 Carotid body PGLs or ICA involvement is a complication risk factor. 4
The most commonly used approach is the transcervical, but depending on location, other procedures such as the trans-mandibular, trans-mastoid, or infratemporal can be used. CN X-involving PGLs need to be approached cervically or through a posterior tear hole. Tympanic PGLs are resected through a low-morbidity trans-canal, microscopic/endoscopic approach. One of the most challenging tumors is the jugular PGL, which can expand to all sensitive structures, such as the ICA, the lower CNs, and the IDS. In these cases, protecting the ICA can lead to substantial technical challenges. Complications of the lower facial nerve or the hypoglossal nerve can affect up to 30% of patients. No consensus on the systematic rerouting of the facial nerve has been reached. Elderly patients with recurrent external acoustic canal hemorrhages may benefit from partial resection. 115
Ideally, endoscopic surgery should be performed in intra-abdominal PGLs. However, several challenging locations, such as the organ of Zuckerkandl and the sympathetic paraganglia, may need open approaches, as incomplete resection of these tumors has a high risk of recurrence and malignancy. 4
Presurgical management
Surgery and anesthesia induction, adrenal venography, and arteriography are potential triggers of massive CMN release.116–118 Respectively, the substantial drop in CMN release postoperatively can also lead to hemodynamic instability with hypotension. 119 As a result, guidelines suggest CMN stabilization with antihypertensive medication 7–14 days before surgery.2,3
Twice-daily nonselective α-adrenergic receptor blockers with dosing titration are the method of choice for controlling CMN levels and managing HTN.2,3 The most widely used options are phenoxybenzamine (irreversible) and doxazosin (reversible). A recent RCT reports no statistically significant difference in total intraoperative time outside targeted BP [SBP<160 mm Hg and mean arterial pressure (MAP) > 60 mm Hg] between the two drugs. Phenoxybenzamine usage, however, seems superior as it is linked to less frequent (p = 0.02), shorter (p < 0.01) SBP spikes, and, most notably, better hemodynamic profile during surgery (p = 0.02) 120 (see Future Directions). A-blockade is unnecessary before IV nonionic contrast administration in patients with suspected or established PPGL.51,116 Finally, biochemically silent or pseudo-silent PPGLs should be managed cautiously, as evidence of the efficacy of α-adrenergic blockade is scarce. However, HTN crisis has been described in case reports.121,122
Additional drugs can be used to manage HTN before the operation. 2 Calcium Channel Blockers are an efficient complementary therapy.123–125 They can be used as monotherapy if orthostatic hypotension is not tolerated during the alpha blockade and in patients with normal BP or slight HTN. 3 Complementary tyrosine hydroxylase inhibitors such as metyrosine can also be used; however, with caution, as they can cause sedation.2,126,127
After successful and sufficient α-adrenergic blockade, administration of β-adrenergic receptor blockers for heart rate (HR) management is feasible.2,3,128 Administration in the inverse order can lead to massive vasoconstriction and HTN crisis. 129 For the same reason, labetalol is contraindicated, as it is a more potent β- than α1-adrenergic receptor blocker. 130 Hypotension prevention from excessive α-adrenergic blockade is also recommended. A high sodium diet and increased fluid intake are recommended, and current guidelines suggest the administration of 1-2L saline 24 h preoperatively to ensure peri- and postoperative normotension, although evidence supporting this is observational. 2
Targeted BP levels are not strictly specified, but current practice suggests a seated blood pressure target <130/80 mmHg and an upright systolic blood pressure > 90 mmHg, as it is associated with a favorable hemodynamic profile.2,3,120 Suggested heart rate targets are 60–70 and 70–80 bpm in seated and upright position, respectively. 3 Telemedicine in preoperative patient monitoring can benefit clinicians and patients (see Future Directions). 131
Metastatic disease
Radionuclide therapy
Milder or moderate cases of metastatic disease can be very responsive to PRRT.2–4,132 Medium activity (200–275 mCi) 123 I-MIBG every 3 months can reach a disease control rate of 63–87% in patients with milder metastatic PPGLs.2,133 The hormonal response rate is reported in 10–71% of patients, while the symptomatic response is 23–90%.133,134 High dose 131 I-MIGB, can deliver higher radioactivity per dose (~ 2500 mCi/mg; 92.5 MBq/μg), thus reaching disease control rate levels of 92% in a phase II trial. 135
177Lu-DOTATATE and 90 Y-DOTATOC are primarily used as therapeutic radionuclides in tumors that show activity in 68 GA-DOTA-SSA.2,136 The low rate of nephrotoxicity reported in 177Lu-DOTATATE makes it the more efficient option. 4 Radiological control has been described in 80% of patients with metastatic SDHB-induced PPGL treated with 177Lu-DOTATATE.95,137 A recent metanalysis of 234 cases with differences in baseline reports a disease control rate of 90%. More prospective evidence will be available (see Future Directions).
Chemotherapy
PPGL is not eligible for 123 I-MIGB/PRRT, and metastatic PPGL with high tumor burden is preferably treated with the Averbuch or cyclophosphamide, vincristine, and dacarbazine (CVD) scheme. More than 40 studies, including clinical trials and meta-analyses, report a disease control rate of 48–100% and progression-free survival (PFS) of 20–40 months for SDHB-related metastatic PPGL, which seem to be the most responsive.138–142 PFS and overall survival (OS) are statistically significant in responders compared to nonresponders in multiple series.2,140,142 Guidelines suggest 6–9 cycles of CVD, but this can be extended up to 20 cycles. 2
Temozolomide (TMZ) monotherapy can be a maintenance therapy after CVD. Recent evidence suggests that TMZ’s effectiveness in treating SDHB-harbored PPGLs and PPGLs, in general, depends on the presence of the methylguanine methyltransferase promoter hypermethylation variant. Overall, the PFS MD can be up to 16.9 months. 138 PARP variants, however, have shown to be potentially chemotherapy-resistant; thus, research on PARP inhibitors is needed to potentiate TMZ efficacy (see Future Directions).51,143 Finally, TMZ can be an acceptable monotherapy alternative to CVD, particularly in patients with milder disease courses or history of comorbidity, and can be used in combination with capecitabine.2,4,47
Targeted therapy
Evidence on targeted therapy for PPGL is currently poor, but inspiration derives from their favorable outcomes in the midgut and pancreatic NETs. Tyrosine kinase inhibitor (TKIs), such as sunitinib or cabozantinib, are treatment options for first-line systemic therapy nonresponse and intolerance.2,4,51,144 Limited retrospective and prospective evidence reports a 57–83% disease control rate and PFS of 13.4 months.145,146 TKI usage is increasingly being investigated in clinical trials (see Future Directions). Everolimus is a mTORC1 inhibitor currently being used for abdominal NETs. The disease control rate from PPGL ranges from 25 to 71% in restricted retrospective literature.147,148 Checkpoint inhibitors have shown similar disease control rates (~75%) to other therapies but low PFS in phase II prospective evidence.149,150 As for cold SSAs, also used in abdominal NETs,151,152 there is no evidence suggesting their usage in PPGL, but the concept seems very promising (see Future Directions). 4
Local ablation and palliative care
Treatment goals for advanced metastatic PPGL include suppressing tumor growth, prolonging survival, and minimizing pain and CMN-related symptoms. Local ablation can lead to local disease control and relief of symptoms and can be considered on a case-by-case basis.4,153 There is no evidence comparing different ablative methods or ablation to non-ablation. A retrospective series of 272 patients with advanced mPPGL who underwent local ablation showed OS and PFS rates of 24.6 and 33.7 years, respectively. The most frequent ablative method was palliative radiotherapy (RT), used in 47% of cases. Other methods included embolization, stereotactic RT, cryoablation, percutaneous ethanol injection, and radiofrequency ablation. 97% of the patients underwent palliative surgery, and 45% survived for over 10 years. 154 Lastly, pain control for bone metastasis, the most common presentation of metastatic disease, should be addressed for a better quality of life (QOL) and increased survival.2–4,51
Follow-up
Determining the risk of recurrence or metastasis
Risk stratification based on characteristics and genetic profile, as suggested by Nölting et al. 4 is crucial for successful and personalized follow-up. Patients should be considered low risk if they have <5 cm tumors and adrenergic biochemical profiles or harbor a low-risk RET variant. HNPGL, high-risk PCC (NA, ⩾5 cm, recurrent, multiple), SDHAF2, SDHC, and SDHD mutations alongside cluster 2 mutations, including high-risk RET variants, should be considered intermediate risk. Finally, mPPGL, sympathetic chain PGL, SDHA, SDHB, FH, HIF2A/ EPAS1, and cluster 3 mutations are considered high-risk cases.
Short-term follow-up
Postsurgical follow-up is vital and independent of metastatic and recurrency potential, as it evaluates the extent of resection. Alongside clinical evaluation, 2 MN, NMN, and 3-MT measurements should be carried out 2–6 weeks postoperatively and compared to baseline values established preoperatively. 3 Measurements in the normal range after surgery suggest resection and no imaging is needed for confirmation. 117 Values below the RI are to be expected when unilateral or bilateral adrenalectomy has been carried out alongside oncotomy. 63 In cases of elevated free metabolites postoperatively or silent tumors preoperatively, anatomical imaging should be considered after 3–6 months. If levels were established preoperatively, biochemically silent tumors can also benefit from pCGA comparison. 2
Long-term follow-up
Guidelines suggest a follow-up of at least 10 years, not 5 years, postoperatively.2,3,155 For intermediate or high-risk patients, counseling on life-long follow-up should be offered, especially in young subjects with germline mutations. 155 Recommendations are summarized in Table 4. As aforementioned, NETest has shown promising efficacy in the early detection of recurrence or metastasis and should be incorporated into follow-up when feasible 79 (see Additional Testing, Future Directions).
Long-term follow-up recommendations based on risk status.

NF1, MAX, TMEM127, and RET.
SDHAF2, SDHC, SDHD, and VHL mutations.
Cluster 1: skull-base to pelvis MRI, contrast-enhanced pulmonary CT Cluster 2: MRIAP. Functional imaging optional.
extensive size/necrosis, high Ki67, and invasion of vasculature and lymphatics: every 6–12 months.
Surveillance screening is indicated in patients with established germline mutations in one of the susceptibility genes, proven or suspected genetic syndromes that feature PPGL, and patients with previous PPGL history (Table 5). 2
Screening recommendations for asymptomatic patients adopted from4.

Skull base to pelvis.
No consensus in alternating between MRI/PET-CT.
Specific intervals ultimately determined by the genotype.
Families of patients diagnosed with germline mutation-induced PPGL can benefit from genetic screening. 2 The minimum age for genetic screening depends highly on the susceptibility gene targeted. Evidence is controversial and no board or multidisciplinary consensus has been reached for SDHx mutations, due to their unique genetic profile (Table 1). The currently suggested minimum age for genetic screening is 5 years in SDHB mutations and 10 years in other SDHx mutations.2,156 Family members undergoing screening for VHL mutations should start at a minimum age of 5 years. 157
Future directions
PPGL knowledge has progressed massively through the last few decades, challenging some misconceptions and improving clinical practice and outcomes. To the best of our knowledge, 48 clinical trials regarding PPGL are active and reported in ClinicalTrials.gov. Of these, six are active but not recruiting, three are not yet recruiting, and six have no status registered. The rest are recruiting patients, with one study recruiting by invitation. Only one study has published results, but they are preliminary.
Seven studies are currently underway and explore the effect of PET/CT in the clinical diagnosis of PPGL. NCT04605848 is centered around using infrared thermography predictors and outcomes of dysglycemia are currently assessed in patients with PPGL (NCT05451134). A clinical trial tries to identify succinate in vivo using 1H-Spectroscopy MRI (NCT04583384).
Four studies are currently underway and are associated with genetics. Arguably, the most compelling examines the impact of the environment on SDHx mutation expression (NCT04481152). Genetics also extends into tumor management, as multiple molecular genomic tools for recurrence and metastasis are surfacing. Nölting et al. 4 detail the importance of genetic classification of tumors in creating a personalized treatment and follow-up plan for PPGL management. The advancements in the field will further increase the existing genotype–phenotype correlations, leading to more predictable and, thus, favorable outcomes. The newer evidence of NETest’s efficacy by Pacak et al. 79 and Modlin et al. 81 should lead to further studies with larger samples and comparisons to relevant markers, as this method could be vital for both diagnosis and follow-up. mRNA testing offers a significant advantage over other markers, as it can detect tumors based on gene expression, ergo, before they become evident biochemically, clinically, or on imaging.
More than half of the studies currently underway are focused on treatment. Four studies are evaluating the efficacy of 131 I-MIGB for refractory and recurrent mPPGL. A phase II study is recruiting patients to assess effectiveness, dosage, and adverse effects of 177Lu-DOTATOC (NCT04276597). This study’s positive results will be vital, as there is limited prospective evidence on the topic. NCT03206060 is also evaluating the effects of 177Lu-DOTATATE on mPPGL. Another study compares the efficacy of 177Lu-DOTATOC administration to 90 Y-DOTATATE administration and equimolar coadministration (NCT04029428).
New crucial evidence is going to be released for drugs as well. NCT05142241 tests the coadministration of PARP inhibitor talazoparib and temozolomide for treatment-resistant cases of PPGL. A prospective phase II study is currently underway, testing the efficacy of anlotinib on mPPGL (NCT05133349). The efficacy and safety of anlotinib are also being evaluated by NCT04860700. Cabozantinib efficacy is being calculated for mPPGL as well (NCT02302833). The CABATEN study assesses the coadministration of cabozantinib and atezolizumab (NCT04400474). Sunitinib, widely used for other NETs, is also being studied (NCT00843037), while we are also waiting for the results of NCT01371201. Axitinib is TKI of the Vascular Endothelial Growth Factor (VEGF) pathway assessed for mPPGL (NCT03839498). The LAMPARA study evaluates the efficacy of cold SSA lanreotide (NCT03839498). In addition, immunotherapy checkpoint inhibitors are currently being assessed in two studies. Targeted therapy through the D2 receptor is also being tested in a phase II study of ONC201 (NCT03034200). Last, a novel vaccine (EO2401) is currently being developed for mPPGL (NCT04187404).
Hopefully, after new data on efficacy, practicing physicians and patients can start benefiting from these great treatment modules. Research will then shift interest to the applications of these options, such as new intervals, dosages, combinations, and so on. Bioinformatics and Artficial Inteligence (AI) will also be incorporated to make therapeutic schemes more efficient. In addition, telemedicine will be gradually implemented for sustainable and comfortable follow-up. The current genomic landscape will expand, and with advances in metabolome and cellular biology, understanding, therapy, and management will become even more personalized. Lenders et al. 2 suggest that recommendation cards will soon be available for clinical use.
Discussion
The two ‘rules’ of the modern era PPGL are: early detection and precise personalization, as established in the guidelines of the Working Group on Endocrine Hypertension of the European Society of Hypertension.2,3 The precision of genetic testing is crucial for accurately establishing tumor behavior patterns in individuals. Personalization is also essential, including evaluating a patient’s symptoms based on the likelihood of PPGL, establishing personalized cutoffs, and tailoring imaging. These improvements have led to earlier detection and more favorable outcomes in both survival and QOL. Multidisciplinary approaches and education for physicians are also vital for effective management. The future of diagnosis, treatment, and follow-up should target the hereditary nature of PPGL for maximal clinical outcomes.
Footnotes
Appendix
Abbreviations.

| PPGL | pheochromocytoma and paraganglioma |
| PCC | pheochromocytoma |
| PGL | paraganglioma |
| HTN | hypertension |
| HRAS | Harvey sarcoma viral oncogene homolog |
| ATRX | alpha-thalassemia/mental retardation syndrome X-linked |
| NGS | next-generation sequencing |
| SGT | sequential genetic testing |
| VUS | variants of unknown significance |
| SDHx | succinate dehydrogenase all variants |
| miRNA | microRNA |
| lncRNA | long circular RNA |
| circRNA | circular RNA |
| TCA cycle | tricarbocylic acid cycle |
| CMN | catecholamine |
| TCA | tricyclic antidepressants |
| NMJ | neuromuscular junction |
| SSRIs | selective serotonin reuptake inhibitors |
| MAOis | monoamine oxidase inhibitors |
| LR+ | positive likelihood ratio |
| LR- | negative likelihood ratio |
| CI | confidence interval |
| BP | blood pressure |
| pCGA | plasma chromogranin A |
| SBP | systolic blood pressure |
| DBP | diastolic blood pressure |
| NA | noradrenergic |
| A | adrenergic |
| TS | takotsubo cardiomyopathy syndrome |
| MI | myocardial infarction |
| CICMPP | Catecholamine-induced cardiomyopathy in pheochromocytoma and paraganglioma |
| T2DM | type 2 diabetes mellitus |
| HN | head and neck |
| SARS | severe acute respiratory syndrome |
| CV | cardiovascular |
| MN | metanephrine |
| NMN | normetanephrine |
| 3-MT | 3-methoxytyramine |
| DA | dopamine |
| PMets | plasma free metanephrines |
| UMets | 24 h urine free metanephrines |
| Cr | creatinine |
| LC-MS/MS | liquid chromatography with tandem mass spectrometry |
| LC-ECD | liquid chromatography with electrochemical detection |
| IA | immunoassay |
| BMI | body mass index |
| SDHB | succinate dehydrogenase subunit B |
| NET | neuroendocrine tumor |
| CT | computed tomography |
| MRI | magnetic resonance imaging |
| MRIAP | MRI of the abdomen and pelvis |
| HU | Hounsfield units |
| MRA | magnetic resonance angiography |
| SPECT | single-photon emission |
| PET | positron emission tomography |
| I-MIGB | iI-metaiodobenzylguanidine |
| PRRT | peptide receptor radionuclide therapy |
| F-FDG | F-fluorodeoxyglucose |
| F-FDOPA | F-fluorodihydroxyphenylalanine |
| FH | fumarate hydratase |
| MAX | MYC-associated factor X |
| SST | somatostatin |
| SSA | somatostatin analog |
| VHL | Von-Hippel Lindau |
| RET | rearranged during transfection |
| NF1 | neurofibromatosis type 1 |
| WMD | weighted mean difference |
| CCB | calcium channel blocker |
| HR | heart rate |
| PFS | progression-free survival |
| OS | overall survival |
| MGMT | methylguanine methyltransferase |
| PARP | poly-ADP ribose polymerase |
| TMZ | temozolomide |
| HSA | high-specific activity |
| TKI | tyrosine kinase inhibitor |
| mTORC1 | mammalian target of rapamycin complex 1 |
| mPPGL | metastatic pheochromoctyoma and paraganglioma |
| RT | radiotherapy |
| QOL | quality of life |
| SDHAF2 | succinate dehydrogenase complex assembly factor 2 |
| SDHA | succinate dehydrogenase subunit A |
| SDHC | succinate dehydrogenase subunit C |
| SDHD | succinate dehydrogenase subunit D |
| HIF2A | hypoxia inducible factor 2α |
| EPAS1 | endothelial PAS domain protein 1 |
| TMEM127 | transmembrane domain protein 127 |
| H&P | history and physical |
| BMP | basic metabolic panel |
Acknowledgements
None.