
Abstract
Pheochromocytomas (PHEOs) and paragangliomas are generally grouped as rare chromaffin cell tumors. The co-occurrence of PHEOs and paragangliomas of the organ of Zuckerkandl (POZ) is extremely rare. The most common symptom of pheochromocytoma-paraganglioma (PPGL) is hypertension, and open surgery is still recommended for the treatment of large PPGLs. Herein, we report a case of a successful simultaneous laparoscopic resection of a large PHEO accompanied by POZ in a 40-year-old man with normal blood pressure. DNA analysis revealed a mutation in the succinate dehydrogenase subunit B in both the PHEO and the POZ. To the best of our knowledge, this is the first report of tumors occurring simultaneously in these two locations. We believe that the co-occurrence of PHEO and POZ is extremely rare, and the possibility of PPGL cannot be ruled out in patients with normal blood pressure. The decision to perform laparoscopic surgery remains questionable for patients with a large PHEO and POZ. In addition, a genetic examination should be performed to identify the existence of PPGL-related inherited syndromes.
Keywords
Introduction
Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare catecholamine-secreting tumors that have an annual incidence of six per million people. 1 Excessive secretion of catecholamines can lead to hypertension, and typical symptoms include headache, palpitation, and hyperhidrosis.2,3 Approximately 80% of cases originate from the adrenal medulla (PHEO), and approximately 20% occur within extra-adrenal sites (PGL). PGL occurs mainly in the thorax, abdomen, and pelvis. Paragangliomas of the organ of Zuckerkandl (POZ)—which consist of paraaortic paraganglia located between the origin of the inferior mesenteric artery or renal artery and the level of the aortic bifurcation—represent approximately half of all PGLs.2,4 Surgical resection is the first-choice treatment for pheochromocytoma-paraganglioma (PPGL or PHEO/PGL). Open surgery is still recommended for PHEOs or POZ with a tumor diameter ≥5 cm, and the security of laparoscopic procedures remains dubious. 1 The co-occurrence of PHEO and POZ is extraordinarily interesting, and no clear guidance exists on the choice of surgical approach. Herein, we report a case of PHEO accompanied by a POZ ≥5 cm in diameter. To the best of our knowledge, this is the first report of tumors occurring simultaneously in these two locations. We successfully performed simultaneous laparoscopic resection in this patient and reviewed the literature on PPGL. The reporting of this study conforms to CARE guidelines. 5
Case report
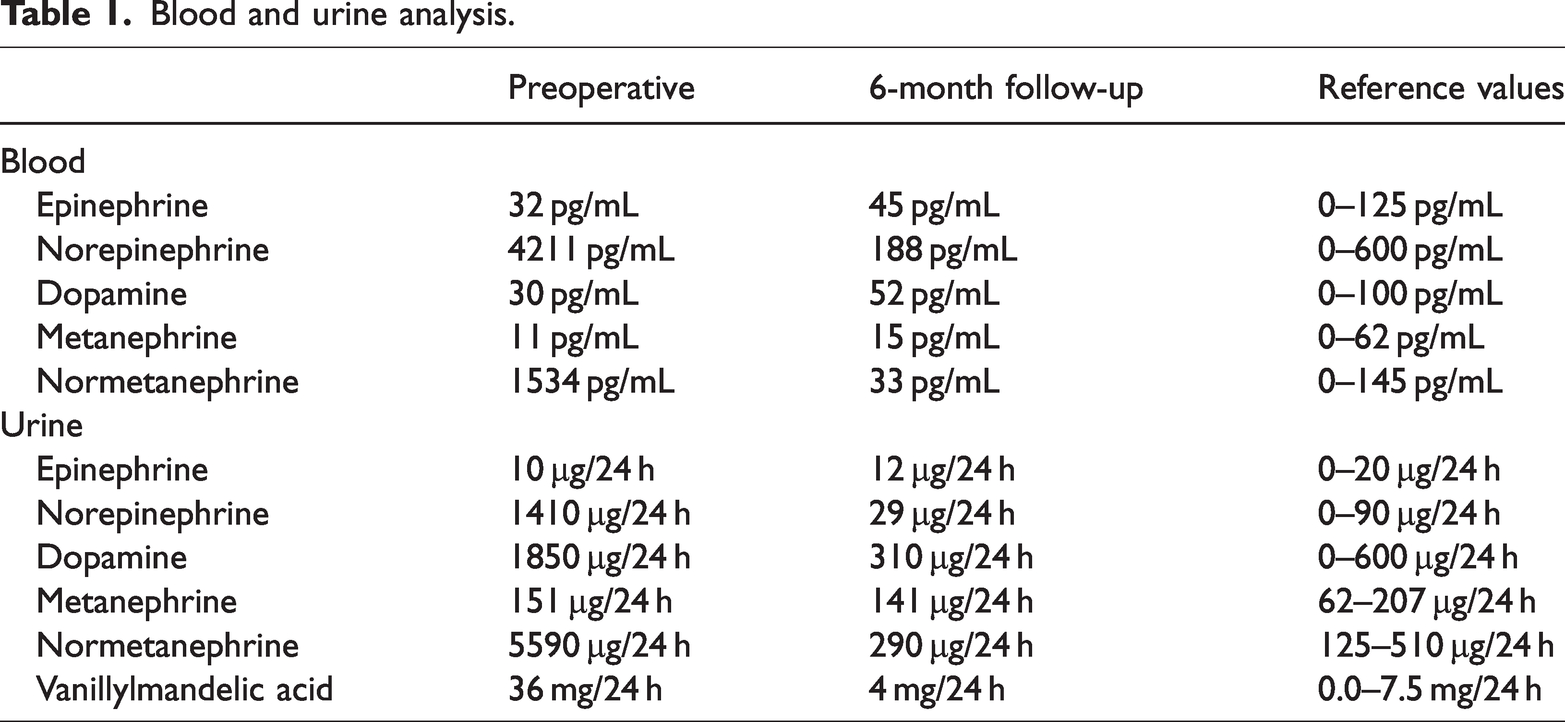
A 40-year-old man presented with a 3-month history of sporadic episodes of palpitations and intermittent diaphoresis. His blood pressure and heart rate were within the normal range, and no abnormalities were observed on physical examination. However, the patient’s serum and urinary catecholamines were elevated (Table 1). A computed tomography (CT) scan (Figure 1) of the abdomen and pelvis showed a rounded mass (5.14 cm × 4.31 cm) in the right adrenal gland and an irregularly shaped mass (7.55 cm × 4.71 cm) at the umbilical level between the retroperitoneal inferior vena cava and the abdominal aorta. The irregularly shaped tumor was located in the Zuckerkandl organ area. However, the patient did not undergo a 123I-metaiodobenzylguanidine (123I-MIBG) scan because of financial considerations. Instead, brain magnetic resonance imaging and neck ultrasonography were performed to exclude the presence of PPGL-related genetic syndrome. Finally, the patient was diagnosed with PHEO and concurrent POZ. The patient was treated with phenoxybenzamine for 2 weeks to prevent intraoperative cardiovascular accidents and postoperative hypotensive shock. After consulting with general medicine and vascular, cardiology, anesthesiology, and other disciplines, the patient underwent laparoscopic surgery and was prepared for possible conversion to open surgery.
Blood and urine analysis.

Computed tomography findings: Right adrenal mass (red arrow) and abdominal mass (black arrow). (a) Transverse plane of right adrenal mass. (b) Transverse plane of abdominal mass and (c) Coronal plane (1) abdominal aorta (2) right renal artery.
After the induction of general anesthesia, the patient was placed in a 70° left lateral decubitus position, and 10-mm, 10-mm, 10-mm, and 5-mm trocars were implanted 2 cm above the umbilicus, under the costal margin, beside the right rectus abdominis, and under the xiphoid process, respectively. Laparoscopy was used for observation following pneumoperitis activation. First, the triangular ligament of the liver was excised, the liver was lifted, and the upper pole of the right kidney was exposed. The right colon was carefully separated and pushed away to expose the right kidney. The descending and horizontal segments of the duodenum were then pushed to expose the inferior vena cava and right renal vein. After further separation, we observed an irregular tumor of approximately 7.55 cm × 4.71 cm in size with a capsule and abundant nutrient vessels at the upper level of the tumor. We separated and ligated the vessels individually and continued to separate downward along the tumor surface. A blood vessel from the abdominal aorta was suspected of supplying the tumor. After clamping using hem-o-lok clamps, the mass in the abdominal cavity was completely removed (Figure 2). The right central adrenal vein was then located and disconnected upward along the lateral side of the inferior vena cava. The mass was separated along the plane between the psoas major muscle and the tumor body. The blood vessels supplying the tumor were carefully dissected. Finally, the adrenal mass was completely removed along its periphery (Figure 2).

Photograph of the gross pathology of the right adrenal and abdominal masses. (d) The abdominal tumor is irregular and was separated into three parts during the operation and (e) The right adrenal mass was well-circumscribed and encapsulated.
The patient’s blood pressure remained stable throughout the surgery, except when the blood pressure soared to 190/110 mmHg when contact was made with the adrenal mass. The operation lasted 130 minutes, and intraoperative blood loss was approximately 100 mL. Postoperative pathological results confirmed the diagnosis of right PHEO accompanied by POZ (Figure 3). Microscopically, the tumor cells were round or polygonal with abundant cytoplasm. Immunohistochemical staining showed loss of succinate dehydrogenase subunit B (SDHB) protein expression and negative chromogranin A. An SDHB mutation was revealed by DNA analysis in both the PHEO and the POZ. After 6 months of follow-up, the patient remained well, and evidence of recurrence was not observed on CT. Serum and urinary catecholamine levels were within the normal range (Table 1).

Microscopic features of the PHEO (f) and POZ (g). The tumor cells were arranged in nests (hematoxylin–eosin staining × 100).
Discussion
PGL mostly occurs in the Zuckerkandl organ, which was first described as an unusual lymph node by Emil Zuckerkandl in 1901.6,7 Our team previously published a case of the successful laparoscopic resection of a large POZ. 4 PHEO was accompanied by POZ, and the diameter of both tumors was >5 cm, which is extremely rare. Laparoscopic surgery in this situation is more complex and necessitates a multidisciplinary team. 8 Patients with PPGL account for 0.2% to 0.6% of the population with hypertension and exhibit paroxysmal (approximately 48%) or persistent (approximately 29%) hypertension and even hypertensive crisis, cardiovascular and cerebrovascular events, noncardiogenic pulmonary edema, and shock.9,10 However, the clinical manifestations of PPGL differ considerably. Regardless of whether the catecholamine level in serum and urine is elevated, clinical signs of PPGL can be minimal or absent; this state is clinically termed “silent PPGL.” Some researchers accept that silent PPGL may be caused by the low number of catecholamine receptors in the cardiovascular framework.11–13 However, misdiagnosis and missed diagnoses are likely to occur in this situation. Therefore, the possibility of PPGL must be considered if paroxysmal signs or symptoms are present. Similarly, our patient had no hypertension other than occasional symptoms of palpitations and hyperhidrosis, which are typical manifestations of PPGL. In addition, the clinical manifestations of POZ may occur in retroperitoneal tumor compression that causes ureteral and small intestinal obstructions.14,15
The diagnosis of PPGL is particularly important because of the negative consequences of misdiagnosis or missed diagnoses. In accordance with the Endocrine Society guideline, the initial biochemical test should include measurements of plasma-free or urinary fractionated metanephrines. 16 123I-MIBG imaging is a more specific type of functional imaging than CT scans. 17 Our patient did not undergo a 123I-MIBG scan examination for financial reasons. However, given the catecholamine spectrum and CT findings, a diagnosis of PHEO accompanied by POZ was established.
PPGL can synthesize and store excess catecholamines. When released, especially during anesthesia induction or tumor manipulation, catecholamines may cause life-threatening cardiovascular complications. 18 Therefore, adequate preoperative management is crucial before PPGL surgery. Reasonable preoperative preparation can significantly reduce perioperative complications. 19 The Endocrinology Society guideline recommends oral administration of phenoxybenzamine or other α-adrenergic receptor blockers for 7 to 14 days and a high-sodium diet and fluid intake before surgery. 16 Currently, the universal view is that laparoscopic resection is an optional procedure for tumors with a diameter of 5 cm or larger and should be performed based on tumor features and the surgeon's experience and technique. 1 Both the PHEO and the POZ had diameters of >5 cm in our patient. Laparoscopic surgery was successfully performed given our surgical team's previous experience and the support of vascular, general, cardiology, anesthesiology, and other specialties. 4
Notably, the patient’s blood pressure soared to 190/110 mmHg during the operation when we touched the adrenal tumor. However, the blood pressure remained stable (fluctuation range: 120/80 mmHg to 130/85 mmHg) when the abdominal tumor was removed. The reason could be that most POZ are often nonfunctional and are called “silent POZ.” 4 Some studies have suggested that POZ may lack tyrosine hydroxylase—the rate-limiting enzyme in catecholamine synthesis—leading to defective synthesis of catecholamines. Alternatively, these tumors may arise from primitive neural crest cells that do not secrete catecholamines. 20 In addition, among the catecholamines secreted by PPGL, epinephrine can only be released by PHEO, with very rare exceptions. This is because epinephrine is converted by norepinephrine under the action of phenylethanolamine N-methyltransferase, which is produced by the adrenal medullary chromaffin cells. Therefore, POZ originating from extra-adrenal sites cannot secrete epinephrine. 21
Previously, 10% of PPGL cases were believed to be inherited. However, a recent study showed that at least 60% of patients have germline or somatic mutations in PPGL-related genes, with at least 30% of these mutations being hereditary.22,23 To date, at least 19 PPGL-related gene mutations have been identified. Because of certain genotype–phenotype correlations, patients with PPGL—especially those who are young or have a family history, patients with bilateral or multiple PPGL, and relatives of PPGL susceptibility gene mutation carriers—should undergo genetic examination. 24 Moreover, germline SDHx, particularly SDHB mutations, are associated with a higher risk of malignancy. 25 SDHB immunohistochemistry is a reliable alternative marker for identifying SDH mutations; thus, all PPGLs should be screened with SDHB immunohistochemistry.2,23 The loss of immunohistochemical expression of SDHB in our patient implied the possibility of an SDH mutation, and further genetic examination confirmed the existence of an SDHB mutation. The patient’s relatives refused to undergo genetic testing despite our recommendation; therefore, we suggested a regular physical examination.
Approximately 10% to 30% of PPGLs relapse or metastasize, and distinguishing between benign and malignant tumors using preoperative biochemistry tests, imaging examinations, and pathological results without metastasis is difficult. 26 The World Health Organization renamed “malignant pheochromocytoma” to “metastatic pheochromocytoma” in 2017, and PGLs were no longer considered benign tumors. 6 In their review of 135 POZ patients, Anuradha Subramanian and Vijay Maker 27 showed that 9% of patients had metastatic disease at the time of initial surgery, 4% had prior resections of PHEOs 6 to 16 years before POZ excision, and 11% returned with recurrence or metastatic disease.
Importantly, the POZ in our patient was removed in three pieces because of severe adhesion during resection that can cause local recurrence; severe adhesion may be a limitation of laparoscopy for large tumors. However, we believe that magnifying imaging of the laparoscopic system can be used to perform more delicate operations and may be more advantageous than open surgery for complex anatomical cases. Therefore, a surgical method for such large multiple tumors must be confirmed with large-sample studies.
In conclusion, co-occurrence of PHEO and POZ is extremely rare, and the possibility of PPGL in patients with normal blood pressure cannot be ruled out. The decision to perform laparoscopic surgery remains questionable for patients with a large PHEO and POZ. In addition, a genetic examination should be performed to identify the existence of PPGL-related inherited syndromes.
Research Data
Research Data for Co-occurrence of pheochromocytoma and paraganglioma of the organ of Zuckerkandl resected simultaneously by laparoscopy: a rare case report and literature review
Research Data for Co-occurrence of pheochromocytoma and paraganglioma of the organ of Zuckerkandl resected simultaneously by laparoscopy: a rare case report and literature review by Zhi Chang, Jiwen Shang, Shushang Yang, Wuyao Qin and Hao Cui in Journal of International Medical Research
Footnotes
Acknowledgement
We thank the patient for allowing this report of their treatment for this rare disease to be published.
Author contributions
Supervision: JS, SY, WQ, and HC. Writing – original draft: ZC. Writing – review and editing: ZC.
Declaration of conflicting interests
The authors declare no conflict of interest in preparing this article.
Ethics statement
Written consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the journal editor.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Shanxi Province Science and Technology Fund (Award No. 201903D321130), which provided financial support for manuscript revision and data collection.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.