
Abstract
Primary adrenal epithelioid sarcoma is a rare lesion of the adrenal gland, and only seven cases have been reported in the domestic and international literature to date. We herein report a case involving a 65-year-old man with primary adrenal epithelioid sarcoma. After being admitted to the hospital with an adrenal mass found on physical examination, the patient underwent laparoscopic right adrenalectomy. Postoperative pathological findings indicated an epithelioid sarcoma (proximal type). Primary adrenal epithelioid sarcoma is a rare malignancy. Diagnosis is challenging and relies on histopathology and immunohistochemical staining.
Keywords
Introduction
Epithelioid sarcoma (ES) is a highly invasive and recurrent malignant mesenchymal tumor with epithelioid morphology; however, it accounts for only <1.0% of soft tissue sarcomas.1,2 ES can occur at any anatomical location and includes proximal type ES (PES) and distal type ES (DES). 3 PES is relatively rare among adrenal tumors, with only seven reported cases to date.4–10 The clinical manifestations, histological morphology, and prognosis differ between PES and DES. We herein report a case involving a patient with ES of the adrenal gland confirmed by pathology and preliminarily discuss the clinical features, diagnosis, treatment, and prognosis of this case based on the relevant literature.
Case report
A 65-year-old man presented to our clinic with a right adrenal mass that had been identified during a physical examination and had been present for >1 month. The patient had no relevant medical history. Following admission, relevant examinations were completed, and blood biochemical examination showed that the plasma adrenocorticotropic hormone concentration was slightly increased for the first time (68.87 pg/mL); other biochemical examinations revealed normal findings. Plain and enhanced computed tomography (CT) of the adrenal gland revealed a round node-like structure with slight hypointensity in the medial branch and body of the right adrenal gland. It had a size of approximately 33 × 40 mm, clear borders, and a CT value of approximately 32 Hounsfield units. The CT scan also showed a peripherally enhancing soft tissue mass (Figure 1). After considering the patient’s examination findings and test results, a nonfunctional adrenal adenoma was highly suspected. After excluding surgical contraindications, laparoscopic right adrenalectomy was performed. During the operation, a rounded mass with a diameter of approximately 35 mm was found in the right adrenal area. The mass was attached to the surrounding tissues. Nevertheless, the operation proceeded smoothly.
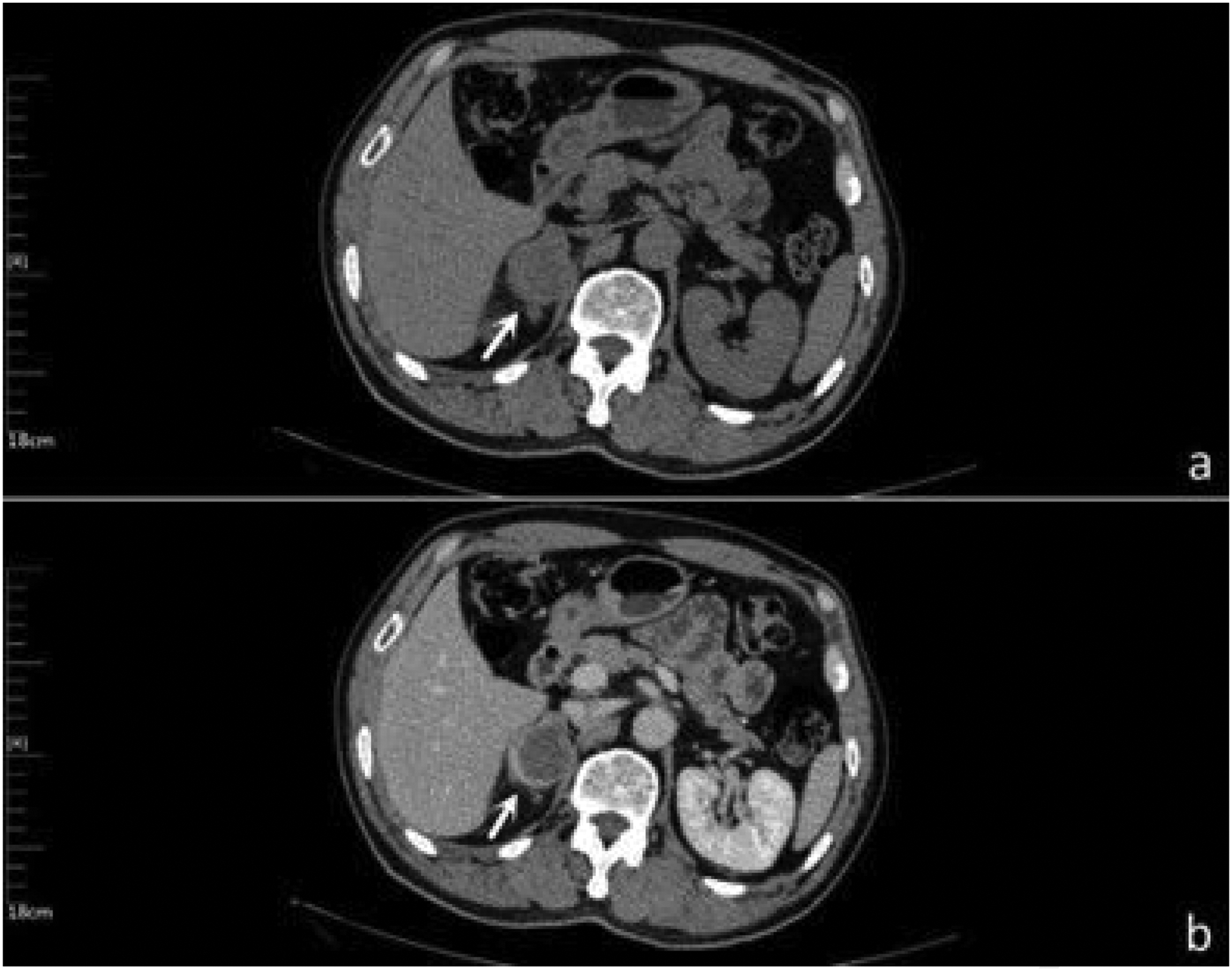
Preoperative imaging data. (a) Plain computed tomography and (b) enhanced computed tomography.
The excised specimen was a round, gray-red, nodular mass with a cystic shape and a wall thickness of 0.1 to 0.3 cm. Histopathological examination revealed map-like necrosis of cancer cells within the mass tissue, and rhabdomyosarcoma-like changes were observed. Additionally, lamellar disc-like clusters and scattered polygonal cells with vesicular nuclei and prominent nucleoli were present (Figure 2). Immunohistochemical (IHC) detection was performed with a BenchMark ULTRA automatic IHC analyzer (Roche Diagnostics, Indianapolis, IN, USA). The IHC results were as follows: inhibin α (−), Syn (−), Ki-67 (+) 20%, CgA (−), EMA (+), S-100 (−), CKp (+), ERG (+), CR (−), CAM5.2 (+), CD31 (−), CD34 (+), INI-1 (−), SMARCA4 (+), and TTF-1 (−) (Figure 3). The postoperative pathological diagnosis was PES.
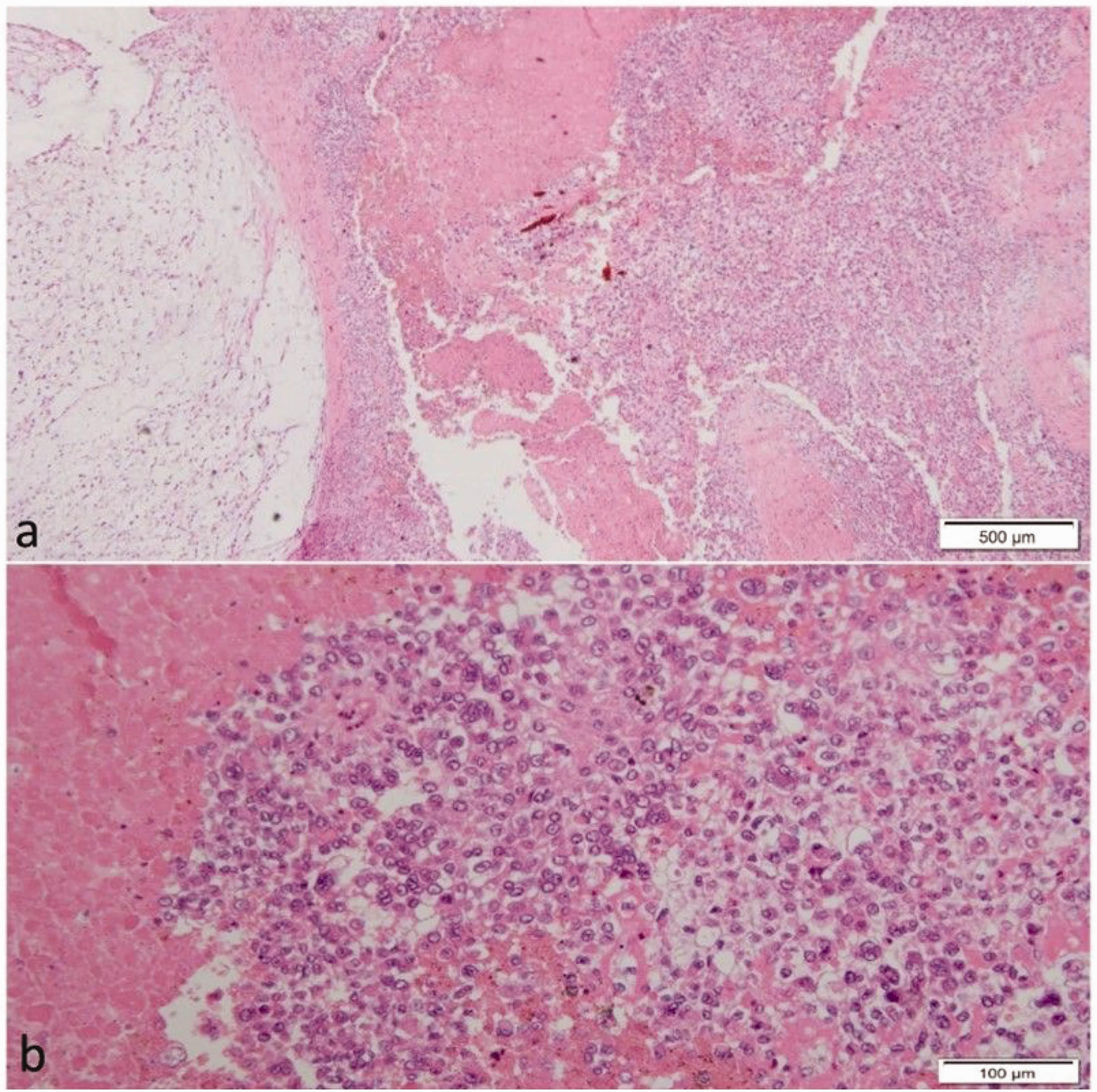
Histopathological images of the tumor tissue with a large number of heterogeneous epithelioid cells and diffuse distribution of tumor cells with rhabdomyoblast-like changes. Hematoxylin–eosin staining. (a) ×40 and (b) ×200.

Immunopathological images, ×100 (EnVision system; Agilent Technologies, Santa Clara, CA, USA). (a) CAM5.2+. (b) CD34+. (c) CKp+. (d) EMA+. (e) RG+ and (f) INI-1−.
Postoperative recovery was fair based on treatment and prognosis. Additional radiotherapy was recommended; however, the patient requested an upper-level hospital visit and refused follow-up, so we were unable to perform the relevant staging for the patient.
The reporting of this study conforms to the CARE guidelines. 11
Discussion
In 1970, Enzinger 3 first introduce the concept of ES as a specific soft tissue malignancy. He named it based on the resemblance of the tumor cells to epithelial cells and their specific epithelial markers. DES usually occurs in the extremities, mainly in the hands and forearms, and is mostly seen in young people aged <35 years. 12 It is more common in male than in female patients and may grow in nodular or multi-nodular forms, frequently with central nodular necrosis and epithelial ulcers. 13 The average age at onset of PES is higher than that of DES (>40 years), and the main sites of onset are the pelvis, perineum, axilla, mediastinum, and genital tract. ES is primarily a soft tissue tumor. Although metastatic hematogenous dissemination can occur with solid organ involvement, primary ES of solid organs is extremely rare. PES and DES can be located in subcutaneous or deep soft tissues; however, PES is more likely to invade deeper tissues, be more aggressive, metastasize, and have a worse prognosis. 3 In this study, a nodular arrangement of spindle and epithelioid cells was observed under microscopic examination of DES, and the granuloma structure demonstrated changes. Overlying dermoid cells were mainly observed under microscopic examination of PES, with obvious atypia and a large nucleolus. Additionally, rhabdomyoma cells were characterized by changes and neoplastic necrosis. Rhabdomyoma and PES show similar characteristics on IHC examination; however, PES behaves more aggressively.3,14,15 Notably, when the adrenal gland becomes infected, necrotic changes can also occur in adrenal gland tissue, and these changes should be interpreted in conjunction with clinical manifestations. 16
ES has no special clinical manifestations and is found incidentally in most cases. Alikhan et al. 4 reported the first cases of primary adrenal ES, and these patients are still being followed up and treated. The main task for differential diagnosis of adrenal ES is to differentiate it from adrenal adenoma and adrenocortical carcinoma. However, it is difficult to distinguish between these neoplasms using imaging and biochemical examination alone. ES is a histopathologically unique malignant mesenchymal tumor with dual mesenchymal and epithelial differentiation. This differentiation enables the tumor to histologically masquerade as benign, malignant, or any reactive manifestation. 17 IHC examination plays a vital role in ES diagnosis; the IHC characteristics are relatively unique in that they strongly express cytokeratin, epithelial membrane antigen, and vimentin. Patients with ES are typically CD34-positive, and INI-1 protein deletion is frequently found.4–10 Therefore, relevant evidence suggests that the most specific finding in the diagnosis is the lack of INI-1 expression in tumor cells, which can also be used to distinguish ES from synovial sarcoma. The current treatment option for PES is lesion resection followed by radiotherapy. However, the outcome of PES is frequently poor because of postoperative recurrence and metastasis, with the lymph nodes and lungs being the most common sites of metastasis.1,3 Therefore, it is necessary to follow up patients with adrenal PES.
Table 1 summarizes the existing literature on adrenal ES. Few studies of adrenal ES have been published to date, and this paper is the seventh relevant case report. Although the diagnosis of adrenal ES mainly relies on IHC examination, some discrepancies in previous reports exist; therefore, further studies are still needed. For example, the low expression of Ki-67 in our patient indicates that her prognosis may be good because relevant studies suggest that Ki-67 is an important marker of cell proliferation. Although there is no standard treatment for adrenal ES, surgical resection with postoperative follow-up is recommended given the aggressiveness and high recurrence rate of ES.
Comparison of cases of primary adrenal epithelioid sarcoma.

EMA, epithelial membrane antigen.
Conclusion
Primary adrenal ES is an extremely rare type of solid organ ES. Clinicians should first rule out whether the lesion is a malignant metastatic tumor. Primary adrenal ES has no specific symptoms in the early stage; it may be associated with back pain or abdominal pain. Imaging also shows no specific changes. A whole-body imaging examination before surgery is recommended to determine whether the tumor is metastatic. The diagnosis depends on postoperative histopathology in combination with IHC examination. Surgical resection of early focal lesions is the only treatment method. The recurrence rate, metastasis rate, and survival rate remain unclear because of the short follow-up time and few reported cases. Because the disease is prone to recurrence and metastasis, imaging examination is very important in postoperative follow-up. Mid- and late-term treatment such as chemotherapy, radiotherapy, targeted therapy, and immunotherapy need to be further discussed and studied.
Footnotes
Acknowledgement
The authors would like to express their gratitude to the staff of the Department of Urology and Pathology for their valuable contributions to this case.
Author contributions
Daocheng Fang and Hui Li contributed to the conception and design of the work, interpretation of the data, drafting of the manuscript and revising it critically for important intellectual content, and the scientific integrity of the work. Yuanyuan Hu contributed to the interpretation of the data and the reading and revising of the manuscript. All authors have read and approved the final manuscript.
Data availability statement
All data underlying the results are available as part of the article, and no additional source data are required.
Declaration of conflicting interests
The authors declare no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Patient consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Statement of ethics
The Medical Ethics Committee of Shanghai Songjiang Central Hospital approved this study (batch number: 2023SQ015).