
Abstract
Objective
Emerging evidence has indicated that excessive reactive oxygen species (ROS) have detrimental effects on osteoarthritis (OA). This study aimed to elucidate the effects of melatonin (MT), an antioxidant indolamine secreted from the pineal gland, on chondrocyte senescence and cartilage degeneration, thereby clarifying the underlying mechanisms of ROS-induced OA pathogenesis.
Design
Hydrogen peroxide (H2O2) was used to induce oxidative stress in rat chondrocytes. ROS levels were evaluated using cytometry and immunofluorescence. Cell viability was detected using the Cell Counting Kit-8 (CCK-8) assay. Western blotting and qPCR (Quantiative Real-Time Polymerase Chain Reaction) were used to examine apoptosis and autophagy. For in vivo experiments, male Sprague-Dawley rats were randomly divided into a sham-operated group, DMM (destabilization of the medial meniscus) surgery group, and surgery groups that received melatonin. Knee joints were collected and stained for histological analysis.
Results
The data demonstrated that melatonin treatment significantly suppressed H2O2-induced matrix degradation and apoptosis, and maintained mitochondrial redox homeostasis. In addition, an enhancement of autophagic flux was observed through western blotting. These findings corresponded with activation of the AMPK/Foxo3 signaling pathways upon melatonin treatment. Histological staining and transmission electron microscopy (TEM) micrographs also demonstrated that melatonin alleviated cartilage ossification and chondrocyte hypertrophy in vivo.
Conclusions
Our results indicated that melatonin protected chondrocytes via mitochondrial redox homeostasis and autophagy. The effects of melatonin on senescence may apply to other age-related diseases. Thus, melatonin may have multiple potential therapeutic applications.
Introduction
Osteoarthritis (OA) is an age-related disease and is characterized by subchondral bone destruction and progressive articular cartilage degradation.1,2 The pathogenesis of OA is not completely understood. Accumulating evidence suggests that oxidative stress plays an essential role in cellular senescence and influences mitochondrial turnover through several mechanisms.3,4 Among these are reactive oxygen species (ROS), which are produced in the mitochondria in response to stress.5,6 Recent studies have demonstrated that excessive accumulation of ROS, caused by mitochondrial dysfunction, can reduce antioxidant defenses. This disrupts homeostasis, causing apoptosis, autophagy, inflammation, and matrix degradation.7-10
The 5’-AMP-activated protein kinase (AMPK), a serine/threonine kinase composed of several catalytic subunits (α, β, and γ), constitutes a major metabolic switch that protects energy homeostasis by matching mitochondrial ATP production to external stressors. 11 Previous studies demonstrated that AMPK activation restored mitochondrial function by maintaining redox status and sustaining mitochondrial membrane potential, which inhibits mitochondria-induced apoptosis. 12 Downstream transcriptional factors dependent on the AMPK signaling axis, such as Nrf2, Sirt1, and Foxo, regulate several genes that reinforce antioxidant defenses.13-15 Moreover, senescent cells respond to oxidative stress through mitophagy, which is the process of selectively clearing long-lived or fragmented mitochondria. 16 Foxo3, a member of the forkhead box transcription factor class O (Foxo) family in mammals, is a master regulator that contributes to antioxidant defenses and the autophagy process.17,18 Foxo3 is ubiquitously expressed. Phosphorylation of Foxo3 causes nuclear translocation and subsequent transcription of its target genes.19-21 Several substances involved in Foxo3 activation are considered potential targets for the treatment of degenerative diseases.22-25 Recent studies have emphasized the emerging influence of AMPK/Foxo-related signaling pathways.
Melatonin (N-acetyl-5-methoxytryptamine) is an indolamine hormone secreted by the pineal gland ( Fig. 1A ), 26 which has been shown to possess potential pharmacological properties including antioxidant and antiapoptotic effects.27-29 The concentrations of melatonin vary dramatically in different tissues at different times. 30 Mitochondria maintain a relatively higher concentration, which ameliorates multi-stress-induced disturbances by maintaining redox homeostasis, membrane potential, membrane dynamics, and mitophagy. Previous studies have demonstrated that co-therapy with melatonin could alleviate the detrimental side effects of other drugs. 31 Although emerging evidence has gradually revealed that the underlying mechanism of melatonin is through AMPK-dependent signaling, 32 few studies have illustrated its influence on cartilage degeneration. The effects of melatonin on apoptosis, autophagy, and chondrocyte biosynthesis remain unclear. Based on the results of recent studies, we examined the protective effects of melatonin against oxidative stress injuries in mammalian primary chondrocytes, focusing on mitochondrial physiology and metabolism. A rat model was used to analyze pathological changes in cartilage in vivo. We aimed to investigate oxidative stress-induced chondrocyte pathologies and provide a potential therapeutic intervention for OA.
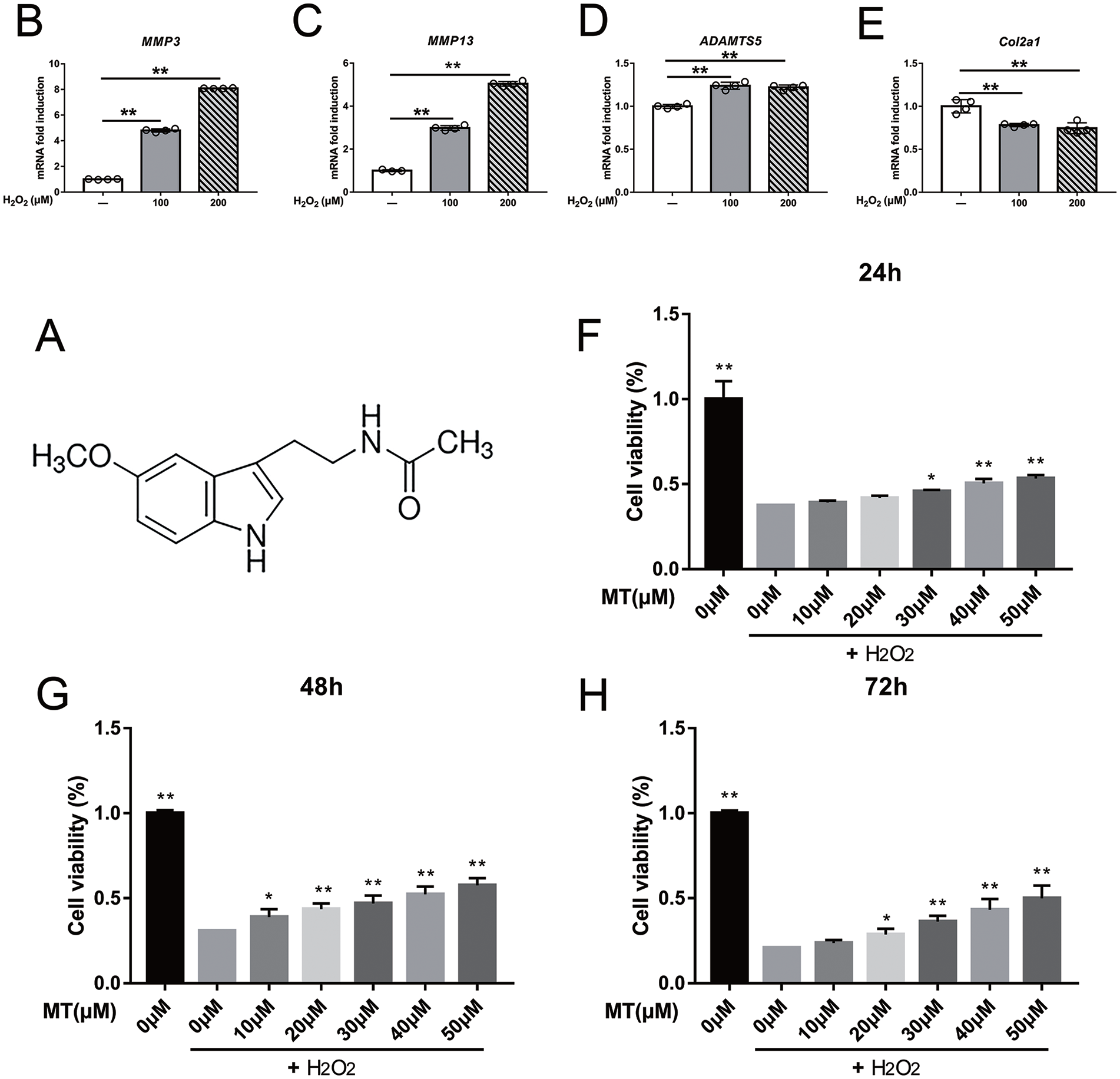
Protection effects of melatonin against H2O2-induced ROS damage on rat chondrocytes, determined by CCK8 assays, and extracellular matrix degradation triggered by H2O2. (A) Chemical structure of Melatonin. (B-E) qPCR results about MMP3, MMP13, ADAMTS5, and Col2a1 mRNA expression after H2O2 (100 μM, 200 μM; 4 h) treatment. MT = melatonin. *P <0.05, **P <0.01, n = 4. (F-H) Cell viability assays of chondrocytes treated with MT for 24, 48, and 72 hours after exposure to 200 μM H2O2 for 4 hours, values are expressed as mean ± standard deviation. *P <0.05, **P <0.01 versus H2O2 alone.
Materials and Methods
Isolation and Culture of Primary Rat Chondrocytes
Primary chondrocytes were isolated from the cartilage tissues obtained from 4-week-old Sprague-Dawley (SD) rats. Cartilage pieces were digested and separated using type II collagenase according to a method described previously. 33 Isolated primary chondrocytes were cultured in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (DMEM/F-12; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco) and antibiotics (Invitrogen, Carlsbad, CA, USA). Cells were seeded and cultured at 37°C in a humidified incubator in a 5% CO2 atmosphere. Subculturing was performed at 70% cell confluence in 10 cm dishes. Cells at the second passage were used for subsequent experiments.
Treatment of Cells
Rat chondrocytes were seeded in 96- and 6-well plates, grown to 60% to 70% confluency, and then incubated in complete growth medium alone or pretreated with melatonin (#M5250, Sigma-Aldrich, St. Louis, MO, USA; 10, 20, 30, 40, and 50 μM) for 2 hours. Hydrogen peroxide (H2O2) (Aladdin, China; 100 μM, 200 μM) treatment for 4 hours was used to model oxidative stress in vitro. An AMPK agonist (AICAR, Beyotime, Shanghai, China; 2.5 mM) and an inhibitor (Compound C, Beyotime; 20 μM) were used to activate or downregulate the AMPK signaling pathway, respectively.
Cell Viability Assays
The chondrocytes were cultured in 96-well plates and treated according to the study design. Cell Counting Kit-8 (CCK; Takara Bio Inc., Tokyo, Japan) was used to detect cell viability. The supernatant was removed after culturing and substituted with a mixed solution containing 100 μL fresh medium and 10 μL CCK-8 reagent, followed by incubation at 37°C for 2 hours. The optical density (OD) at 450 nm was measured using an automatic microplate reader (Infinite M200 Pro, Tecan, Switzerland).
Quantitative Real-Time Polymerase Chain Reaction
After treatment, total cellular RNA was extracted from the chondrocytes using TRIzol reagent (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The RNA extracts were reverse transcribed into cDNA using PrimerScript RT Master Mix (Takara Bio Inc.). RNA quantity and purity were measured using a NanoDrop 2000 (Thermo Scientific). The reaction solution consisted of 2 μg RNA extraction, 4 μL of reagent mixture with cDNA, and H2O2 in a total volume of 20 μL. Primers were designed using Primer Premier (version 6.0, PREMIER Biosoft, Palo Alto, CA, USA) and validated online using Primer-BLAST (National Center for Biotechnology Information, Bethesda, MA, USA). The primer sequences are listed in Table 1. qPCR was performed using a Quantstudio 6 Flex Real-Time PCR system (Thermo Scientific) under the following conditions: 50 °C for 2 minutes, 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 minute. The comparative Ct (2-△△Ct) method was used to detect the relative expression levels of the target genes.
List of Primers Used in Quantitative Real-Time Polymerase Chain Reaction.
Intercellular ROS Detection
Intercellular ROS levels were analyzed using an ROS assay kit (Beyotime). Primary chondrocytes were pretreated with different concentrations of melatonin and then stimulated with or without H2O2 (200 μM) for 4 hours. After 24 hours, rat chondrocytes were incubated with 10 μM dichloro-dihydro-fluorescein diacetate (DCFH-DA) at 37 °C for 20 minutes in the dark. ROS levels were estimated using flow cytometry by measuring the fluorescence emissions at 525 nm, due to the oxidation of DCFH-DA to dichlorofluorescein (DCF) at 488 nm. Representative fluorescence micrographs of ROS were obtained using a Leica DMi8 confocal microscope (Leica Camera AG, Wetzlar, Germany).
TUNEL Staining and Immunofluorescence
Samples were fixed with 4% formalin for 20 minutes and permeabilized with 0.2% Triton X-100 (w/v) in PBS for 10 minutes. A terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining kit (Beyotime) was used to detect cell death. Sections were incubated with a TUNEL reagent mixture for 60 minutes and rinsed with PBS, three times for 5 minutes each, according to the manufacturer’s instructions. For immunofluorescent testing, sections were blocked with 5% bovine serum albumin (BSA) for 30 minutes at room temperature after permeabilization and then incubated with the primary antibody against microtubule-associated protein 1 light chain 3 (LC3) (1:200 dilution, Sigma-Aldrich) at 4 °C overnight. This was followed by incubation with the secondary antibody for 2 hours in the dark. Micrographs were captured using a Leica DMi8 confocal microscope (Leica Camera AG, Wetzlar, Germany).
Transmission Electron Microscopy (TEM)
Cartilage was extracted from the knee joints, washed twice with PBS, and immediately immersed in fresh 2.5% glutaraldehyde for 24 hours at 4 °C, then fixed with 1% osmium tetroxide for 2 hours at 24 °C. Fixed samples were dehydrated in a gradient ethanol series, embedded with epoxy resin, and cut into ultrathin sections. Autophagic microstructures of chondrocytes were observed using a TEM (Hitachi, Tokyo, Japan).
Immunoblot Analysis
Primary chondrocytes were harvested and lysed using a RIPA (Radio Immunoprecipitation Assay) lysis buffer (Beyotime) containing phosphatase and a protease cocktail. Protein lysates were prepared and their concentrations were determined using a bicinchoninic acid (BCA) protein assay kit (Beyotime). Equivalent quantities of proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF, Merck Millipore, China) membranes and blocked with tris-buffered saline and Tween 20 (TBST) containing 5% BSA for 1.5 hours at 24 °C. Proteins were probed with specific primary antibodies and an appropriate fluorescence-conjugated secondary antibody. Protein bands were visualized using a western assay system (Odyssey V3.0, LI-COR Biosciences, Lincoln, NE, USA). The primary antibodies used in this study were as follows: β-actin (1:1000, #4970s, Cell Signaling Technology, Danvers, MA, USA), LC3 (1:1000, #L7543, Sigma-Aldrich), SQSTM1/P62 (1:1000, #ab109012, Abcam, Cambridge, UK), Foxo3a (1:1000, #2497s, Cell Signaling Technology), p-Foxo3a (1:1000, #8174s, Cell Signaling Technology), p-AMPK (1:1000, #2535s, Cell Signaling Technology), AMPK (1:1000, #5832s, Cell Signaling Technology), Bcl-2 (1:1000, #ab196495, Abcam), Bax (1:1000, #14796s, Cell Signaling Technology), cleaved caspase 3 (1:1000, #AF7022, Affinity Biosciences Ltd, Cincinnati, OH), and cleaved PARP (1:1000, #94885s, Cell Signaling Technology).
Animals
Forty 8-week-old male SD rats were purchased from Sino-British Sippr/BK Lab Animal Ltd (Shanghai, China). The animals were fed under appropriate temperature and light conditions in a specific-pathogen-free (SPF) environment with adequate food and water supplies. OA was induced by destabilization of the medial meniscus (DMM) of the right knee. The rats were randomly divided into four groups: the sham-operated group, the DMM surgery group, the surgery group that underwent low-dose treatment with melatonin (15 mg/kg), and the surgery group that underwent high-dose treatment with melatonin (30 mg/kg). The day after DMM surgery, the rats in the treatment groups received melatonin diluted in saline water by intraperitoneal injection every other day. Normal weight-bearing and activity were permitted for all the animals. At 8 weeks after surgery, the rats were euthanized, and the knee joints were collected. All procedures were approved by the Animal Use and Care Committee of Shanghai Jiao tong University and were conducted following the “Guide for Care and Use of Laboratory Animals” by the National Institutes for Health.
Histology and Immunohistochemistry (IHC)
The rat joint tissues were fixed with formaldehyde, decalcified in 10% ethylenediaminetetraacetic acid (EDTA) solution, dehydrated in ethanol, embedded in paraffin, and then sliced into 5 μm sections. The sections were dewaxed, rehydrated, and subjected to hematoxylin and eosin (H&E) and Safranin-O staining. For IHC, the sections were treated with 3% H2O2, blocked with goat serum, and probed with the primary antibody against LC3 (1:200, #L7543, Sigma-Aldrich) and anti-8-hydroxy-2’-deoxyguanosine (8-OH-dG) (#ab48508, Abcam). Subsequently, the sections were incubated with a biotin-linked secondary antibody and counterstained with hematoxylin. Images were acquired using a microscope (Olympus, Tokyo, Japan).
Statistical Analysis
All data are expressed as the mean ± standard deviation, and the differences among groups were assessed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for comparisons between multiple groups. Statistical analysis was performed using GraphPad Prism software (version 5.0, San Diego, CA, USA). Statistical significance was set at a P value < 0.05. All experiments were performed in triplicate or duplicate.
Results
Melatonin Protects Chondrocytes from Oxidative Toxicity Induced by H2O2
Primary chondrocytes were exposed to H2O2 (100 μM, 200 μM) for 4 hours to assess ROS-induced cell toxicity. PCR analysis was performed to assess the expression of genes that influence cartilage matrix degradation and synthesis. As shown in Fig. 1B-D , H2O2 stimulation significantly increased the mRNA levels of MMP3 and MMP13, with ADAMTS5 showing a relatively slight upregulation compared with the control group. Col2a1 expression was not significantly suppressed by H2O2 ( Fig. 1E ). To detect anti-oxidative effects, primary chondrocytes were pretreated with melatonin at different concentrations (10, 20, 30, 40, and 50 μM) for 2 hours before exposure to H2O2 (200 μM). Cell viability was tested at 24, 48, and 72 hours ( Fig. 1F-H ). These results indicated that melatonin markedly attenuated H2O2-induced cellular toxicity in chondrocytes.
H2O2-Induced Oxidative Stress Suppressed by Melatonin
Several antioxidant factors were assessed using qPCR. These include MnSOD, CAT, GSH-Px1, and GSH-Px4 ( Fig. 2A ). Notably, the expression of these factors was upregulated by melatonin in a dose-dependent manner, whereas H2O2 triggered a compensatory increase. Intracellular ROS was measured by flow cytometry using DCFH-DA ( Fig. 2B ), a fluorescent probe that freely crosses the cell membranes, that is oxidized by intracellular ROS to generate brightly fluorescent DCF. H2O2 prominently increased fluorescence in chondrocytes compared with the control cells, while pretreatment with melatonin eliminated excess ROS and attenuated fluorescence intensity. Micrographs of the experiment reflected these differences in intracellular ROS ( Fig. 2C ). Mitochondrial depolarization caused by ROS is regarded as an early event in apoptosis. This is supported by previous studies that illustrated an impairment of mitochondrial membrane potential. To evaluate ΔΨm, an indicator of mitochondrial membrane potential, we probed chondrocytes with JC-1 dye. Control cells stained with JC-1 exhibited distinct red fluorescence due to the aggregation of high concentrations of dye monomers in mitochondrial matrices. Dye monomers gathered around the cellular matrix and produced green fluorescence, which the dye exhibits when in a low concentration, indicating a reduction in ΔΨm. Compared with the control group, H2O2 decreased the JC-1 aggregate/monomer ratio. Notably, pretreatment with melatonin rescued the H2O2-induced ΔΨm reduction ( Fig. 2D ).
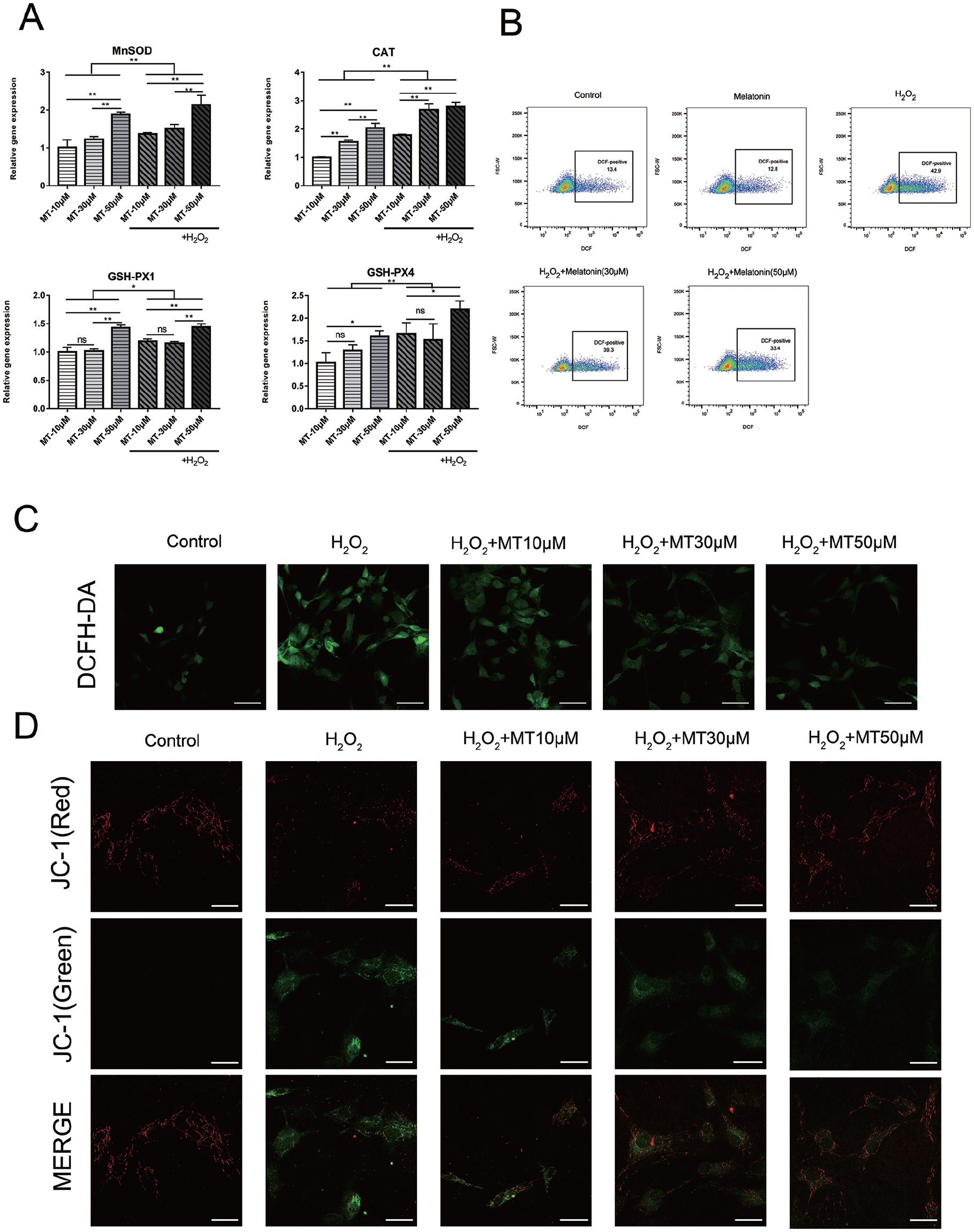
Melatonin attenuates the H2O2-induced mitochondrial dysfunction and suppresses oxidative stress in rat primary chondrocytes. (A) Chondrocytes were pretreated with melatonin (10 μM, 30 μM, 50 μM) for 2 hours, then incubated with H2O2 (200 μM) for 4 hours. mRNA expression levels of MnSOD, CAT, GSH-PX1, and GSH-PX4. Data represent mean ± standard deviation. *P < 0.05, **P < 0.01, n = 4. (B) Chondrocytes were pretreated with melatonin (30 μM, 50 μM) for 2 hours, then incubated with H2O2 (200 μM) for 4 hours. Intracellular ROS was tested by flow cytometry. (C) Representative fluorescence micrographs of ROS obtained by fluorescence microscopy. (D) Fluorescence measurement of mitochondrial membrane potential by JC-1. Scale bar = 30 μm. DCFH-DA = dichloro-dihydro-fluorescein diacetate; ROS = reactive oxygen species.
Melatonin Attenuates H2O2-Induced Apoptosis
Significant reductions in ΔΨm result in mitochondrial outer membrane permeabilization (MOMP), which triggers the release of apoptosis-related intermembrane proteins. Pro-apoptotic BAX is an upstream trigger of caspase activation. This study demonstrated a higher expression of BAX in the H2O2- treated group. This was reflected in a parallel downregulation of Bcl-2, which plays a critical role in regulating BAX recruitment ( Fig. 3A-C ). Furthermore, downstream apoptotic proteins, such as caspase-3 and PARP, were also investigated. Pretreatment with melatonin decreased the expression of caspase-3 and PARP post H2O2 stimulation, indicating an attenuation of apoptosis ( Fig. 3D-E ). The results of TUNEL staining also proved that H2O2-induced oxidative stress dramatically increased apoptosis. Melatonin-treated chondrocytes showed fewer apoptotic cells ( Fig. 3F ).
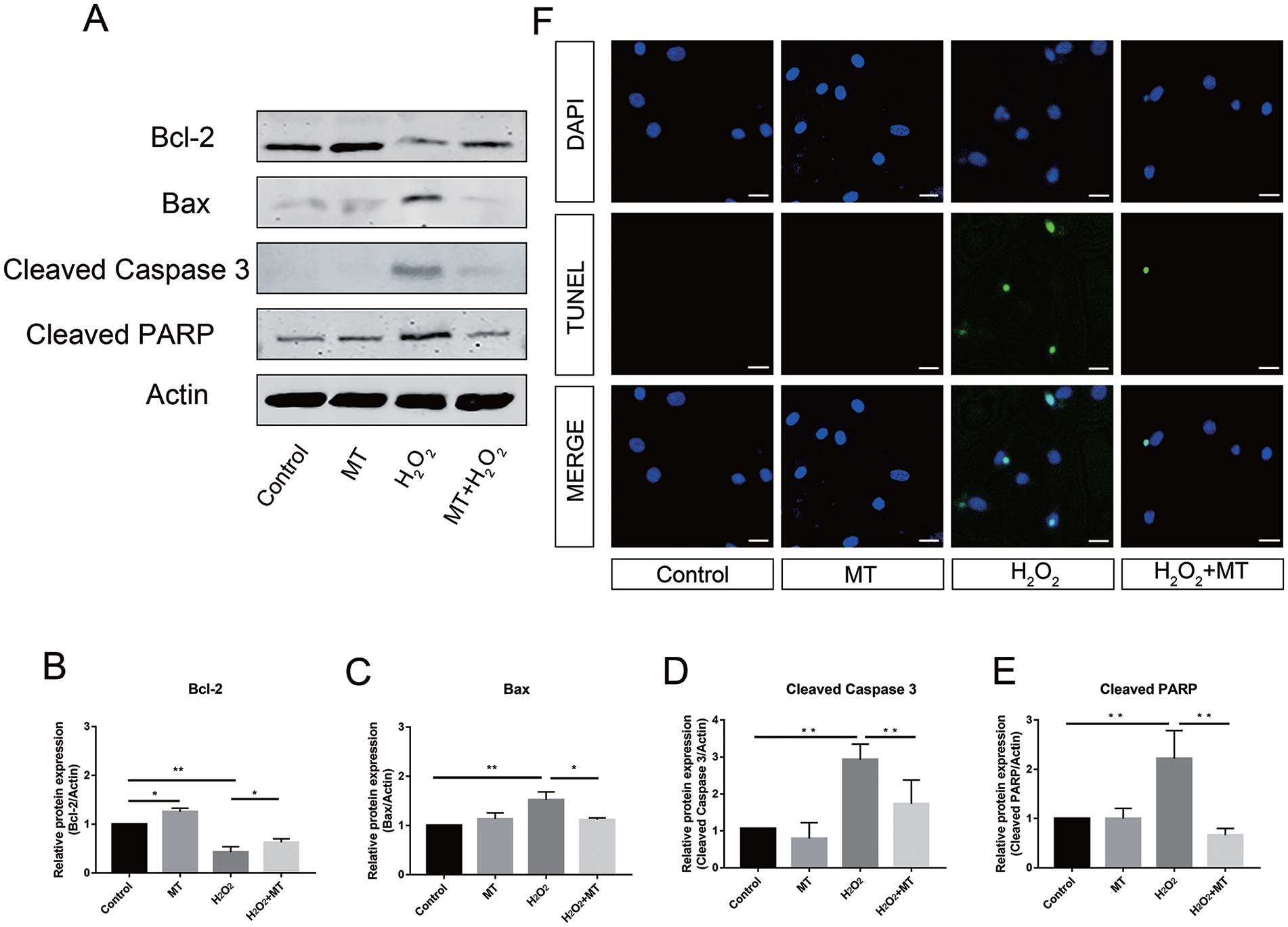
Protective effect of melatonin on H2O2-induced mitochondria-mediated apoptosis in rat chondrocytes. Chondrocytes were pretreated with melatonin (50 μM) for 2 hours, then incubated with H2O2 (200 μM) for 4 hours. (A) Western blotting bands of Bcl-2, Bax, Cleaved Caspase 3, and Cleaved PARP (Poly ADP-Ribose Polymerase). Protein quantitation of Bcl-2 (B), Bax (C), cleaved caspase-3 (D), and cleaved PARP (E). Data represent mean ± standard deviation. *P <0.05,**P <0.01. (F) TUNEL analysis of apoptotic chondrocytes, nuclei were stained with DAPI (4‘,6-Diamidino-2-Phenylindole). Scale bar = 20 μm. TUNEL = transferase dUTP nick end labeling.
Melatonin Upregulates Cell Autophagy
Mitophagy plays a vital role in metabolic homeostasis and LC3 is a key factor in autophagy. Normally, the baseline expression of LC3 is relatively low and stable. Increased LC3 levels were observed in the groups pretreated with melatonin, which was confirmed by fluorescence staining that showed increased LC3 expression in those chondrocytes ( Fig. 4A ). The ratio of LC3-II to LC3-I (LC3-II/LC3-I) is an important indicator of autophagic flux, where LC3-I is converted to LC3-II. SQSTM1/p62 is an autophagy adaptor that links the ubiquitin pathway with the autophagy pathway and is degraded in the process. Here, we found that melatonin increased the LC3-ll/LC3-I ratio and upregulated the total amount of LC3 protein, while the p62 expression level was reduced ( Fig. 4B ). Next, we observed autophagy activation in the rat model of DMM. TEM showed that the numbers of autophagosomes and autolysosomes remained limited in the sham-operated and DMM groups. Notably, hypertrophic chondrocytes in the DMM model secreted a substantial number of fat granules. Fusions of autophagic organelles were seen in the melatonin-treated group ( Fig. 4C ). Taken together, melatonin upregulated chondrocyte autophagic activity under oxidative stress both in vitro and in vivo.
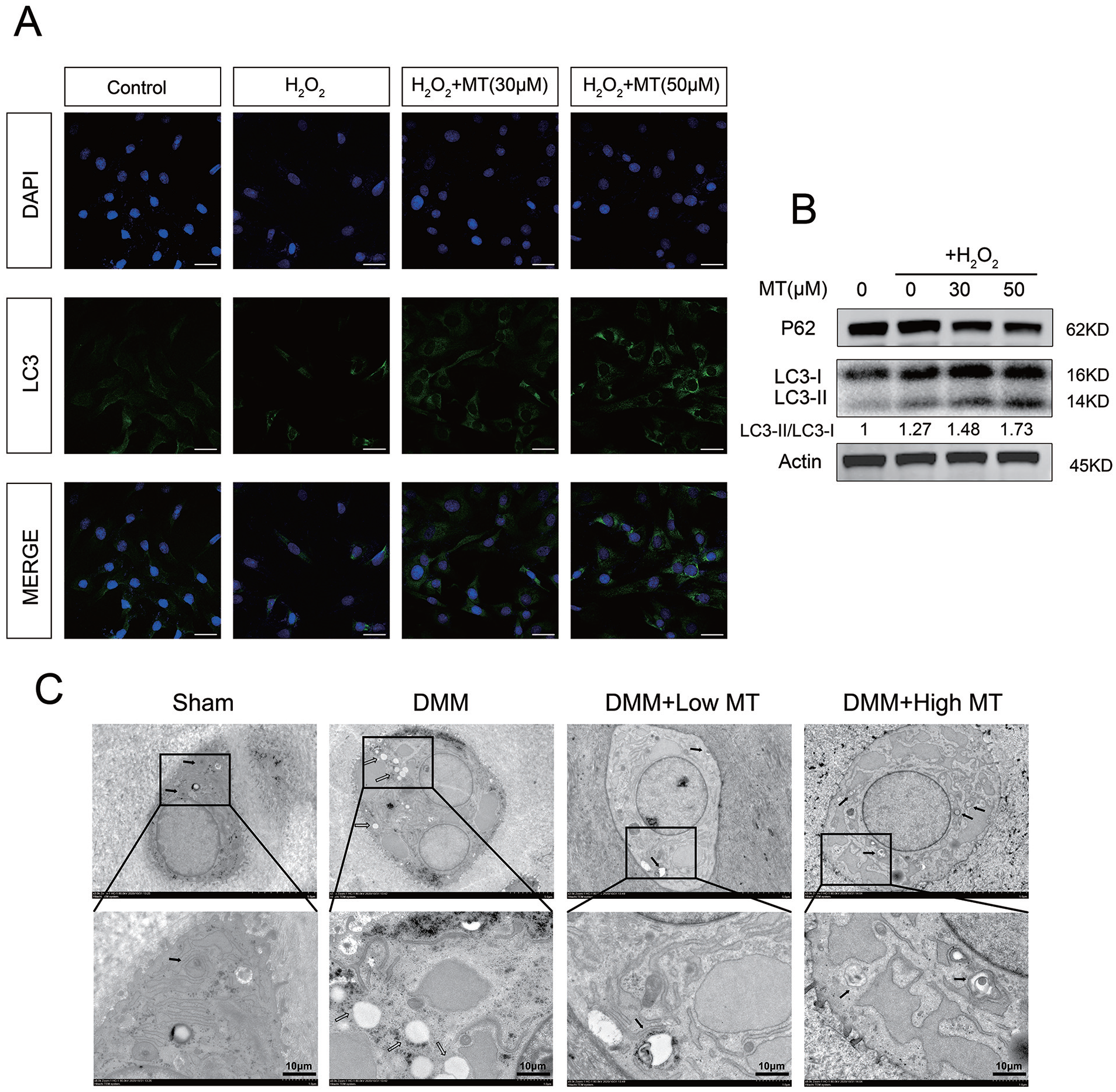
Melatonin regulate autophagic flux in rat chondrocytes. (A) Representative immunofluorescence staining of LC3 in rat chondrocytes. Scale bar = 30 μm. (B) Protein expression of P62 and LC3 in rat chondrocytes obtained by Western blot analysis. (C) TEM images of knee-joint cartilage at 8 weeks after the operation. Low MT and High MT represent for 15 mg/kg and 30 mg/kg, respectively. DMM group presented more hypertrophic lipid droplet compared with control group. In the melatonin treatment model, the autolysosome fusions improved as the dose of melatonin increased. Autophagosome and autolysosome were identified by black arrows, while empty arrows marked abnormal lipid droplet. Original magnification 8,000x, scale bar = 10 μm. TEM = Transmission Electron Microscopy; MT = melatonin; DMM = destabilization of the medial meniscus.
Activation of the AMPK/Foxo3 Signaling Pathways by Melatonin
Next, we investigated the effects of melatonin on chondrocyte signaling pathways, such as the AMPK system, which influences stress response and anti-aging effects. In the presence of melatonin, p-AMPK and p-Foxo3 were upregulated. To confirm the importance of AMPK activity in Foxo3 phosphorylation, we used Compound C, a chemical inhibitor of AMPK that does not significantly inhibit structurally related protein kinases, such as protein kinase A and protein kinase C. Expression of p-AMPK and p-Foxo3 was dramatically downregulated due to the inhibitory effect of Compound C. We also found that chondrocytes subjected to continuous co-treatment with AICAR and melatonin demonstrated a co-activation effect, demonstrated by increased protein expression of both p-AMPK and p-Foxo3 ( Fig. 5A-E ). These data suggest that melatonin likely plays an essential role in AMPK activation beyond just inducing Foxo3 transactivation, and that its transcriptional signaling activity may also be involved in autophagy.
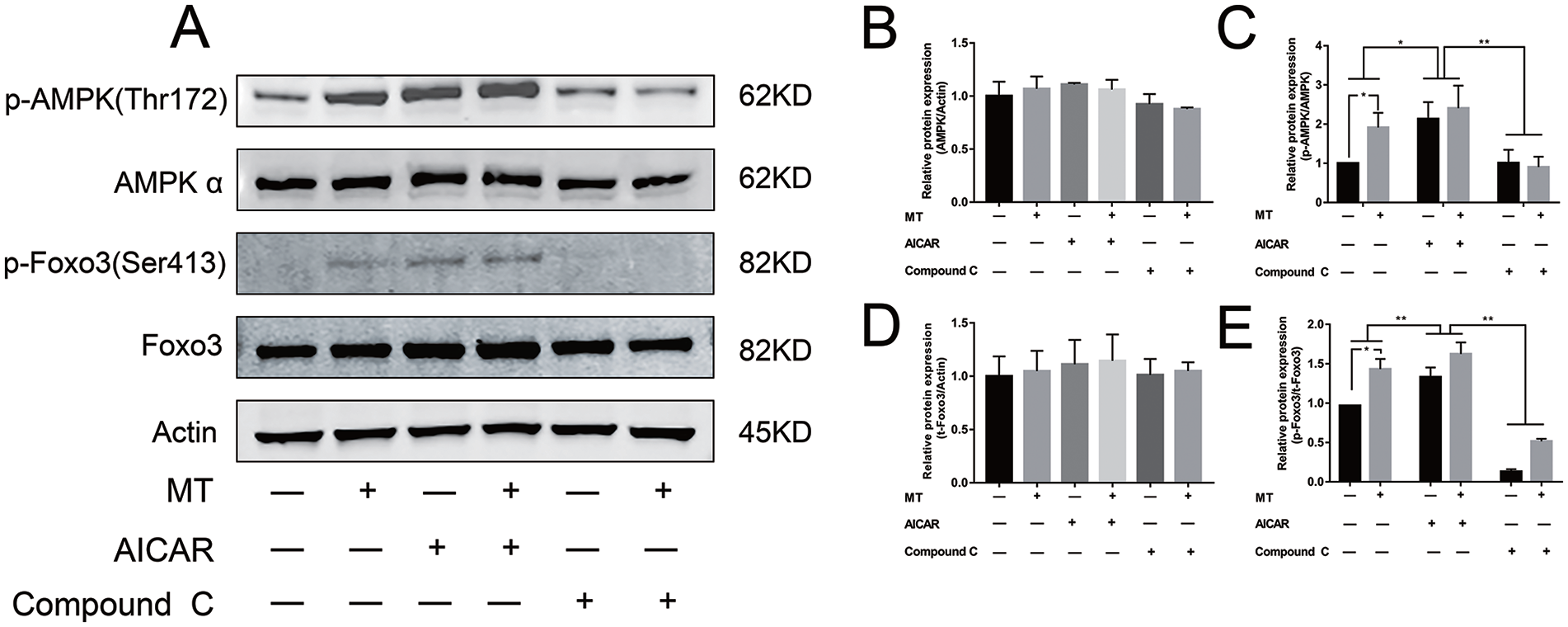
Melatonin regulates AMPK/Foxo3 pathways. (A) Protein expression of p-AMPK, AMPK, p-Foxo3, and total-Foxo3. β-actin served as loading control. Chondrocytes were pretreated with AICAR (5-Aminoimidazole-4-Carboxamide 1-β-D-Ribofuranoside) (2.5 mM) or Compound C (20 μM) for 2 hours followed with continuous exposure. Then a co-treatment of melatonin (50 μM) was performed together. Quantitative results for AMPK (B), p-AMPK (C), t-Foxo3 (D), and p-Foxo3 (E) based on protein bands. Data represent mean ± standard deviation. *P <0.05, **P <0.01. AMPK = 5’-AMP-activated protein kinase.
Melatonin Attenuates Cartilage Degeneration in a Rat DMM Model
To further evaluate the therapeutic potential of melatonin for OA, we designed a DMM model using SD rats. Eight weeks after surgery, the knee joints were harvested for subsequent histologic staining and IHC. The histological morphologies of the knee joints from all groups were analyzed. The cartilage thickness was reduced and the surface boundaries were more damaged in the DMM group compared with the sham-operated group. Moreover, the DMM group showed a decreased number of chondrocytes in the cartilage layer ( Fig. 6A-D ). Safranin-O staining demonstrated that melatonin restored cartilage matrix; an extracellular matrix that stained red indicated the formation of hyaline cartilage rather than fibrocartilage ( Fig. 6E-H ). Furthermore, the number of LC3-positive cells was significantly higher in the melatonin-treated group, as demonstrated by the IHC ( Fig. 6I-L ). Staining of 8-OH-dG also demonstrated alleviated oxidative damage in melatonin-treated groups ( Fig. 6M-P ). The statistical analysis confirmed that melatonin treatment may have a positive effect on OA.
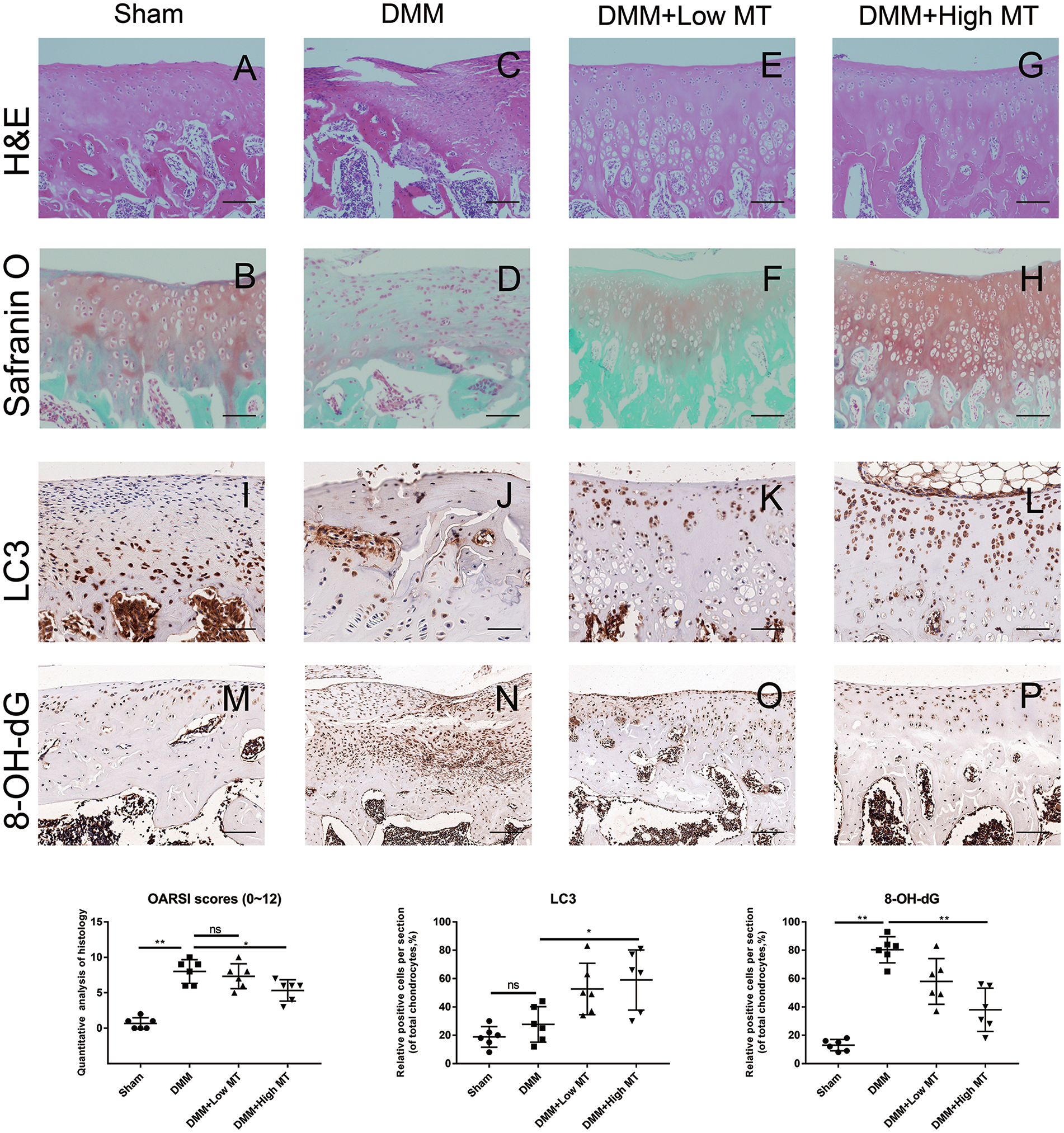
Melatonin inhibits cartilage degeneration in a rat model. (A, C, E, and G) Hematoxylin and eosin staining of knee-joint cartilage in sham group and model groups of SD rats at 8 weeks posttreatment. Low MT and High MT represent for 15 mg/kg and 30 mg/kg, respectively. (B, D, F, and H) Saffron solid green staining of knee-joint cartilage in sham and model groups at 8 weeks posttreatment. (I-L) IHC staining of LC3 in cartilage at 8 weeks after the operation. (M-P) 8-OH-dG staining of cartilage at 8 weeks after the operation. Original magnification 100x, scale bar = 100 μm. Osteoarthritis Research Society International (OARSI) scores and IHC staining were statistically analyzed. Data represent mean ± standard deviation. *P < 0.05, **P < 0.01. SD = Sprague-Dawley; IHC = histology and immunohistochemistry; MT = melatonin; DMM = destabilization of the medial meniscus.
Discussion
Hyaline cartilage is avascular, which means that nutrients and metabolic wastes are exchanged less freely. Evidence from in vitro and in vivo studies suggests that oxidative stress and mitochondrial dysfunction in chondrocytes play a pivotal role in OA. Accumulated ROS directly contribute to extracellular matrix degradation and intercellular apoptosis. H2O2, a main source of ROS, is widely involved in redox homeostasis. Oxidative stress induced by H2O2 can be enhanced in pathological environments, resulting in OA progression. Due to the avascular nature of cartilage, we hypothesized that certain endogenous substances in the vascular circulation must play a critical role in aging resistance, energy maintenance, and apoptosis regulation. Melatonin is an indoleamine secreted by the pineal gland. The potential therapeutic effects of melatonin on degenerative disease have attracted attention in recent years, and its antioxidant properties have been speculated to attenuate OA exacerbations.32,34 Here, we studied the protective effects of melatonin on cartilage. In vitro tests demonstrated that melatonin improved chondrocyte viability against H2O2-induced oxidative stress. Apoptosis induced by H2O2 and hyaline cartilage matrix degradation were both attenuated by melatonin. Mitochondrial dysfunction was also alleviated by melatonin, as indicated by the increased expression of genes correlated with ROS-elimination and restoration of mitochondrial membrane potential. An OA model using SD rats was also designed to evaluate melatonin’s protective effect in vivo ( Fig. 7 ).
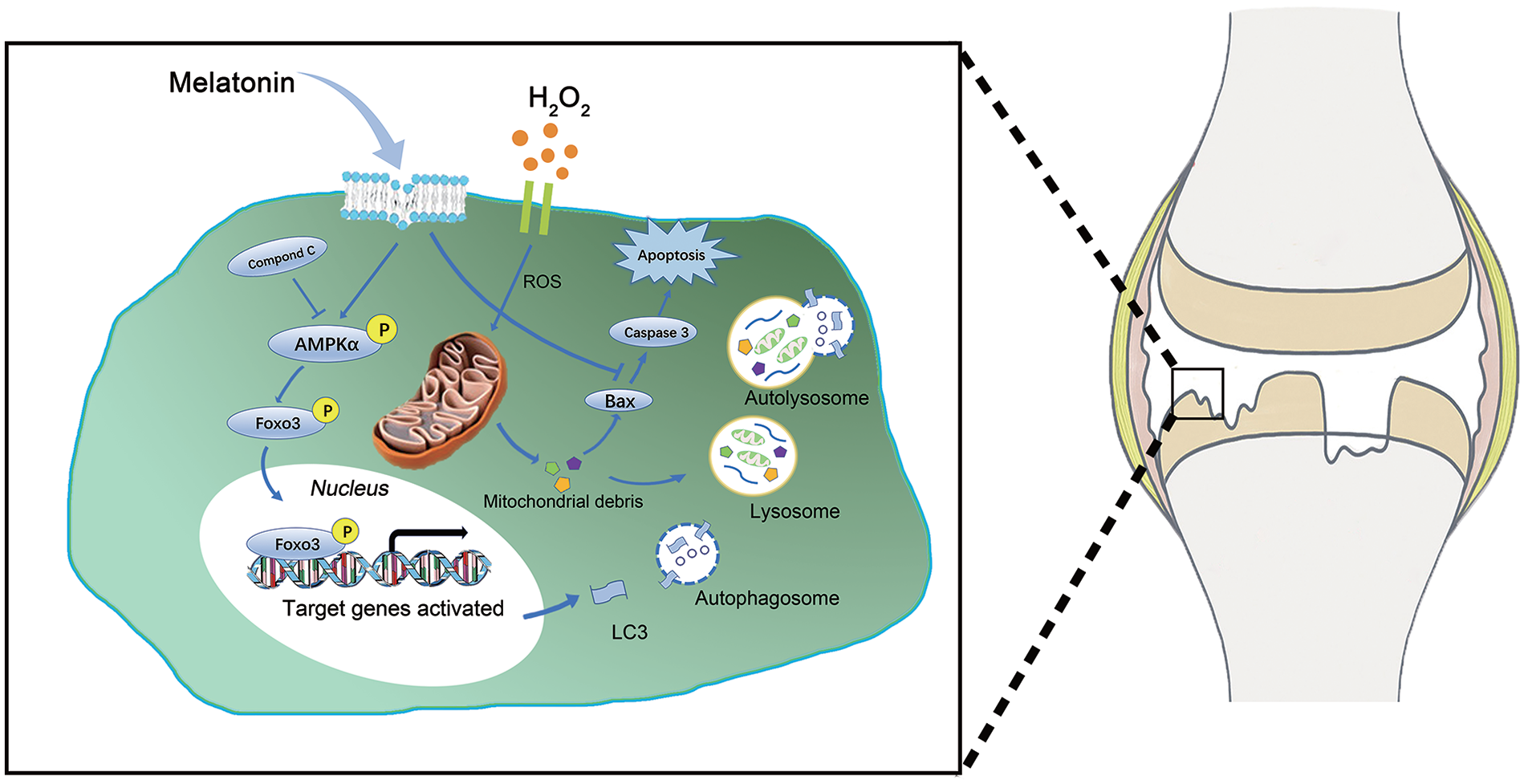
Schematic illustration of the mechanism of melatonin protective effects on rat chondrocytes against oxidative stress induced by H2O2. ROS = reactive oxygen species.
Activation of the AMPK signaling pathway was reported to be involved in ROS regulation.35 -37 The AMPK pathway acts as a fuel gauge by switching on catabolic pathways for ATP generation when energy is insufficient. The transcription factor Foxo3 is phosphorylated via the AMPK pathway, leading to transcription of downstream genes. During our research on melatonin, we identified the AMPK-Foxo3 axis as a potential pathway by which melatonin ameliorates OA progression. Our data demonstrated that Foxo3 activation was significantly promoted by melatonin via the AMPK pathway. Furthermore, co-activation of AICAR, an AMPK agonist, enhanced this process, while Compound C, an AMPK antagonist, reversed it. According to previous studies, Foxo3 increases the expression of autophagic genes, such as ATG7, by binding to its promoters. ATG7 is directly involved in the synthesis of autophagic proteins and the fusion of autolysosomes. 38 Additional evidence supporting Foxo3’s role in autophagy included the upregulation of LC3 and enhanced autophagic flux in vitro. In vivo evidence came from the melatonin-treated rats. In this group, ultrathin sections of joint cartilage were sliced and histological staining was performed. Imaging of these sections using TEM revealed a clear increase in the number of autophagosomes and autolysosomes in the melatonin-treated groups. IHC staining of LC3 also proved that autophagy was active in the restored hyaline cartilage tissues.
This study had some limitations. First, the effects of melatonin on H2O2-induced oxidative stress have not been fully elucidated. The underlying mechanisms of Foxo3 and its target genes are incompletely understood. In addition, the relationship between Foxo3 and autophagy in cartilage remains to be clarified. Last, only DMM rat models, a classical surgical approach, were used to simulate OA degradation. Inflammation caused by destructive mechanical stressors may not perfectly simulate oxidative injury in chondrocytes in vivo.
In summary, we propose that by anti-apoptotic effects and activation of autophagy, melatonin treatment attenuates OA progression in both cellular and animal models. Moreover, the beneficial effects of melatonin and the role of AMPK/Foxo3 signaling pathways in the alleviation of senescence may be common to other age-related diseases. This study highlights new potential therapeutic approaches using melatonin and/or AMPK-Foxo3-targeting drugs in a clinical application.
Footnotes
Author Contributions
Zhaoxun Chen and Chen Zhao contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Acknowledgments and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: National Nature Science Foundation of China, Award Number: 81871791.
Ethical Approval
All animal procedures were approved by the Ethics Committee of Shanghai Ninth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine (approval number A92-1).