
Abstract
Purpose of Review
Mitochondria are recognized to be one of the most important organelles in chondrocytes for their role in triphosphate (ATP) generation through aerobic phosphorylation. Mitochondria also participate in many intracellular processes involving modulating reactive oxygen species (ROS), responding to instantaneous hypoxia stress, regulating cytoplasmic transport of calcium ion, and directing mitophagy to maintain the homeostasis of individual chondrocytes.
Designs
To summarize the specific role of mitochondria in chondrocytes, we screened related papers in PubMed database and the search strategy is ((mitochondria) AND (chondrocyte)) AND (English [Language]). The articles published in the past 5 years were included and 130 papers were studied.
Results
In recent years, the integrity of mitochondrial structure has been regarded as a prerequisite for normal chondrocyte survival and defect in mitochondrial function has been found in cartilage-related diseases, such as osteoarthritis (OA) and rheumatoid arthritis (RA). However, the understanding of mitochondria in cartilage is still largely limited. The mechanism on how the changes in mitochondrial structure and function directly lead to the occurrence and development of cartilage-related diseases remains to be elusive.
Conclusion
This review aims to summarize the role of mitochondria in chondrocytes under the physiological and pathological changes from ATP generation, calcium homeostasis, redox regulation, mitophagy modulation, mitochondria biogenesis to immune response activation. The enhanced understanding of molecular mechanisms in mitochondria might offer some new cues for cartilage remodeling and pathological intervention.
Introduction
Mitochondria synthesize adenosine triphosphate (ATP) to provide energy for cells including chondrocytes through the tricarboxylic acid cycle (TCA, also called Krebs cycle or citric acid cycle) and oxidative phosphorylation (OXPHOS). 1 In addition to producing ATP, mitochondria are also involved in several important cellular physiological activities, such as the production and regulation of reactive oxygen species (ROS), 2 the sensing and adjustment of hypoxia state by hypoxia-inducible factor-1 (HIF-1), 3 the involvement of mitochondria-mediated apoptosis,4,5 and the accommodation of intracellular transport of calcium ions. 6 Besides, mitochondria are essential for the pathogenesis and progression of diseases. Recent reports have shown that mitochondrial damage may play a significant role in the pathological mechanism of osteoarthritis (OA), 7 rheumatoid arthritis (RA), 8 intervertebral disk degeneration (IVDD), and post-traumatic osteoarthritis (PTOA).9,10
Cartilage is a special structure composed of dense extracellular matrix (ECM) and highly differentiated cells called chondrocytes ( Fig. 1A ). ECM plays a crucial role in regulating chondrocyte functions. 11 In terms of composition, ECM is mainly composed of collagen which accounts for 75% of the dry weight of cartilage tissue, and proteoglycan (PG) accounts for 20% to 30% of dry weight.12,13 Among collagen, the proportion of type II collagen is about 85%. 14 More than 90% of the composition in PG comes from aggrecan (ACAN). 15 Chondrocytes are localized in a relatively low-oxygen environment. Compared with 13% to 17% oxygen tension in arterial blood, the oxygen tension in the surface of cartilage is about 5% to 7%. Normal cartilage tissue exists an oxygen gradient microenvironment and the chondrocytes in the deepest regions could only receive about 1% oxygen.16,17 Both glycolysis and OXPHOS exist in chondrocytes and a large proportion of chondrocyte energy requirements are met via glycolysis, rather than OXPHOS ( Fig. 1B ), and OXPHOS only provide about 25% of total ATP in chondrocytes. Nonetheless, OXPHOS’s net yield is about 36 molecules of ATP per molecule of glucose. 18 Compared with OXPHOS, glycolysis is less efficient that each molecule of glucose produces only 2 molecules of ATP. 1 Hence, OXPHOS plays a crucial role in chondrocyte metabolism. Mitochondrial dysfunction could break the balance between the glycolysis and OXPHOS, reducing ATP production substantially. 19
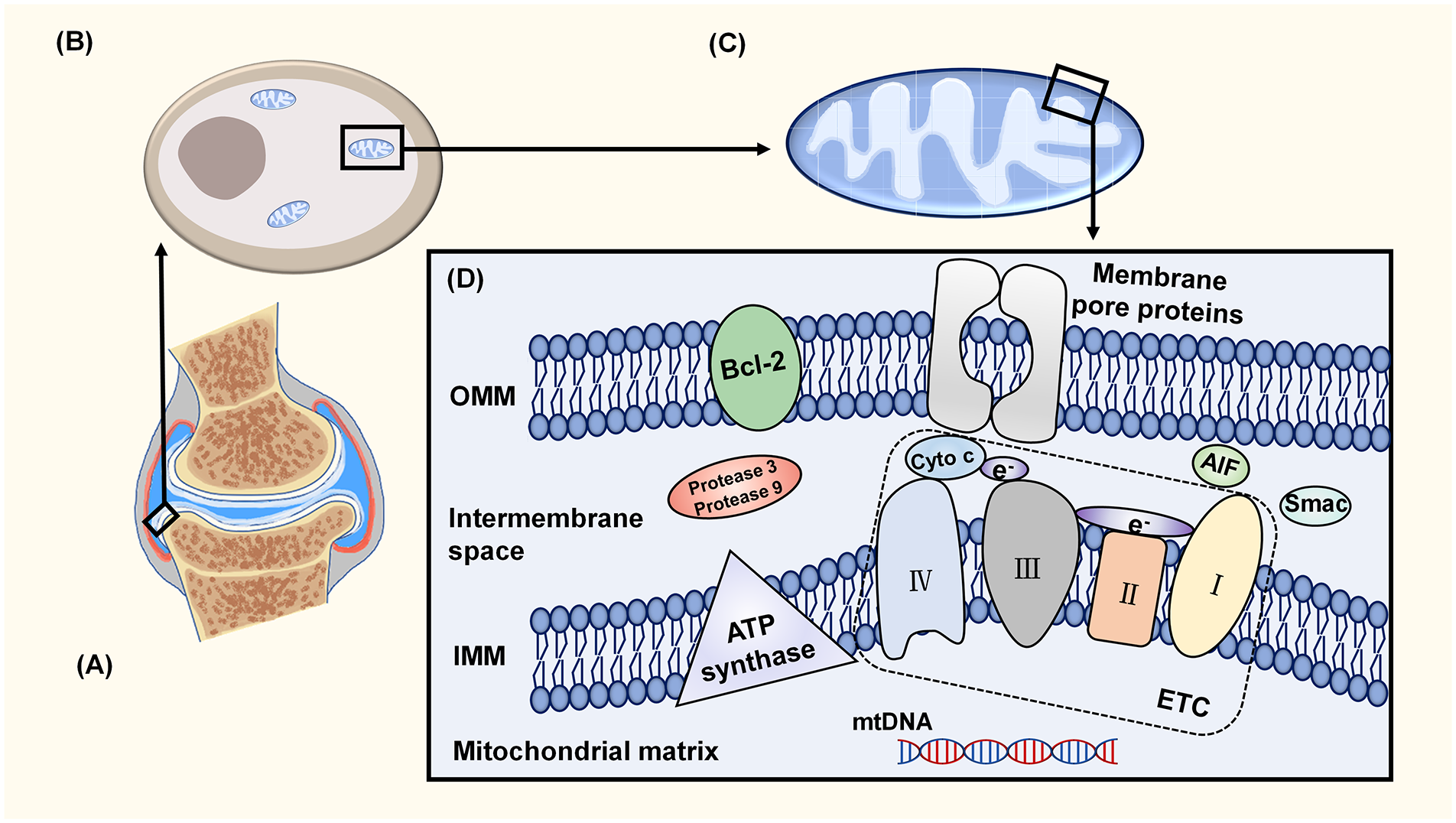
Mitochondrial structure in chondrocytes. (
Mitochondria play a vital role in chondrocyte metabolism because mitochondria not only provide the necessary ATP for chondrocytes, but also participate in the regulation of cellular processes such as redox homeostasis regulation, 20 cellular calcium ions (Ca2+) balance, 21 chondrocyte death signals,9,22 and mitochondrial biogenesis. 23 ATP production in chondrocytes is heavily dependent on the OXPHOS. Electron transport chain (ETC) establishes a proton gradient across the inner mitochondrial membrane (IMM) and generates mitochondrial membrane potential (ΔΨm), driving complex V (also called ATP synthase) to generate ATP. 24 ROS is a sensitive signaling component of cell physiology, which acts to balance cellular redox reaction. Excessive ROS activates signaling pathways such as the insulin phosphatidylinositol-3-kinase-protein kinase B (PI-3K/Akt) and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK). Activated signaling pathways promote cartilage degradation by inhibiting matrix synthesis, cell migration, and growth factor biological activity.25-27 Mitochondria maintain a relatively low ROS level by highly efficient antioxidant system including superoxide dismutase (SOD), α-tocopherol or α-tocopheroxyl radical, glutathione peroxidase (GPX), and so on.28,29 Ion Ca2+ is taken up by mitochondria when cytoplasmic Ca2+ is elevated. The Ca2+ absorbed by mitochondria is used to regulate mitochondrial activity and stimulate ATP synthesis. Ca2+ could regulate 4 mitochondrial enzymes (glycerol phosphate dehydrogenase [GPDH], pyruvate dehydrogenase complex [PDH], isocitrate dehydrogenase [IDH], and oxoglutarate dehydrogenase [OGDH]) in the process of TCA, resulting in an increase of nicotinamide adenine dinucleotide (NADH). Subsequently, increased NADH induces higher respiratory chain activity and enhances mitochondrial ATP production. 30 Apoptosis pathways consist of mitochondrial-mediated autophagy and general autophagy. 4 Mitochondrial-mediated way, also called mitophagy, is a vital cellular apoptosis pathway in chondrocytes. Cellular damage accompanies with a depolarization of ΔΨm. And the change of ΔΨm leads to a release of pro-apoptotic cytokines such as B-cell leukemia/lymphoma-2 (Bcl-2) family members and cytochrome c (Cyto c). Then, Cyto c binds to cytoplasmic scaffolding protein, apoptotic protease activating factor-1 (Apaf-1). The complex of Cyto c and Apaf-1 activates pro-caspase 9 and induces the opening of mitochondrial-mediated apoptosis pathway. 31 The homeostasis of mitochondrial biogenesis is maintained by a mitochondrial quality control system (MQC). MQC mainly preserves functional mitochondria by controlling division and fusion and dynamically removes nonfunctional mitochondria. 32 Mitochondrial dysfunction leads to abnormal ATP synthesis, 33 excessive ROS, 34 matrix calcification, 35 chondrocyte death, 36 mitochondrial biogenesis deficiency, 37 and overactive innate immune responses. 38 Mitochondrial respiratory chain (MRC) dysfunction and ΔΨm depolarization result in a decrease of ATP production as well as an increase in ROS. Ultimately, these pathological changes induce the occurrence of apoptosis. 31 The inhibition of mitochondrial respiration could also promote the mineralization of chondrocytes mediated by matrix vesicles (MVs), induce microcalcification, and cause chondrocyte dysfunction. 39 At the same time, mitochondrial dysfunction leads to an imbalance between fission and fusion that breaks the homeostasis of mitochondrial quality. 40 Mitochondrial biogenesis deficiency decreases the phosphorylation of adenosine 5′-monophosphate-activated protein kinase (AMPK). The decline of AMPK phosphorylation further leads to the depression of NAD+-dependent deacetylase sirtuin-1 (SIRT-1) and the mitochondrial biogenesis master regulator proliferator-activated receptor γ coactivator 1α (PGC-1α) as well as the increase of acetylated PGC-1α. These changes accelerate pro-catabolic responses in chondrocytes. 41 Under the stimulation of hypoxia, mitochondrial dysfunction could also alter the bioenergetics of cells and promote immune cells to be in an abnormally hyper-metabolically active state. 38 Herein, we review the physiological function and pathological roles of mitochondria in chondrocytes to enhance our understanding of mitochondrial mechanisms and highlight the importance roles of mitochondria in cartilage-related diseases.
Structure of Mitochondria in Chondrocytes
Mitochondria are organelles that contain double-membrane structures ( Fig. 1C ). The outer mitochondrial membrane (OMM), the IMM, the space between the mitochondrial membranes, and the mitochondrial matrix are the 4 structural areas of mitochondria ( Fig. 1D ). 42 Membrane pore proteins and Bcl-2 family proteins are located in the OMM. Membrane pore proteins control the exchange of substances in mitochondria. 43 Bcl-2 proteins not only regulate mitochondrial apoptosis, but also involve in mitochondrial dynamics. Bcl-2 family causes the loss of OMM integrity and induces mitochondria to participate in cell apoptosis and this process is always accompanied by the fission of mitochondria. 44 Complex I (also called NADH dehydrogenase), II (also called succinate dehydrogenase), III (also called cytochrome c oxidoreductase), and IV (also called cytochrome c oxidase) of the ETC and complex V are located in the IMM. IMM is the main location for ATP synthesis. IMM folds to form a large number of cristae, which enlarges the surface area of IMM and enhances the ability of ATP generation.17,45 The space between the mitochondrial membranes mainly contains some proapoptotic factors such as Cyto c, apoptosis-inducing factor (AIF), second mitochondrial-derived activator of caspases (Smac), and proteases 3 and 9. Some genetic material is located in the mitochondrial matrix, such as mitochondrial deoxyribonucleic acid (mtDNA) and energy metabolism–related proteins. 46
Most mitochondrial functions rely on the formation of ΔΨm. ΔΨm is formed when the concentration of protons is asymmetrically distributed on the 2 sides of the IMM. 17 The maintenance of ΔΨm is related to many factors. First of all, the structure of the ETC must be intact. MRC complexes, coenzyme Q (CoQ, also called ubiquinone), and Cyto c are indispensable components in the ETC. 42 A complete ETC establishes a proton gradient across the IMM, also called polarization, which generates the ΔΨm and drives the complex V to produce ATP. 24 Researchers have confirmed that using specific inhibitors or RNA interference technology to block the activity of the MRC could lead to a depolarization in ΔΨm.47-50 For example, tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which are 2 main cytokines involved in cartilage degradation, affect the generation of ΔΨm by reducing the activity of MRC complex I. 51 However, the level of intracellular ATP is also important for the maintenance of ΔΨm. The intracellular ATP and ΔΨm are interdependent. Cells use ATP to maintain membrane potential, and the production of ATP also requires mitochondria to provide membrane potential within a normal range. 52
The integrity of the mitochondrial structure is the basis of mitochondrial biological functions. In several mitochondrial-mediated diseases, researchers observed a loss of folded cristae structures and a depolarization of ΔΨm, suggesting a dysfunction of mitochondria. 45 The inhibition of MRC complexes III and V results in an overexpression of pro-inflammatory mediators and an increase of ROS production. Excessive pro-inflammatory mediators and ROS further activate nuclear factor kappa B (NF-κB) signaling pathway which involved in the pathogenesis of OA. 53
Hence, any defect in the ETC or the process of ATP synthesis may induce mitochondrial dysfunction, leading to a dysregulation of normal physiological functions in chondrocytes.
Physiological Role of Mitochondria in Chondrocytes
In recent years, accumulating evidence indicate that mitochondria may play a significant role in the physiology of chondrocytes. 54 The functions of mitochondria in chondrocytes include the production of ATP, the maintenance of calcium homeostasis, the regulation of redox state, and the involvement of mitophagy. A proper mitochondria physiology is a precondition for cell survival. Furthermore, the normal physiological functions of mitochondria are also essential to maintain the stability of the organismal internal environment. 4 Chondrocytes can respond to stress and metabolic variations by changing the structure, number, and function of mitochondria. 42 Therefore, a precise mechanism is needed to explain the connection between mitochondrial homeostasis and chondrocytes homeostasis.
Energy Generation
Mitochondria are indispensable energy-producing organelles in chondrocytes. And OXPHOS plays an important role in synthesizing ATP and providing energy ( Fig. 2A ).55,56 Enzyme complexes involved in electron transport in the respiratory chain are located on the IMM. 57 ATP is synthesized from adenosine diphosphate (ADP) via ATP synthase which is located in the end of the ETC. 58 During the process of OXPHOS, 4 ETC complexes (respiratory chain complexes I-IV) and complex V coordinate to generate ATP and provide energy for chondrocytes. Articular cartilage is an avascular, nonlymphatic, and noninnervated tissue. 31 The chondrocytes in cartilage are surrounded by a relatively low-oxygen extracellular environment.17,59 Traditionally, chondrocytes are considered to be highly glycolytic cells. And the ATP provided by OXPHOS only accounts for a small part of the total energy in chondrocytes. 59 However, reports have confirmed that OXPHOS is a kind of more effective way to synthesize ATP. And OXPHOS plays a crucial role in the energy metabolism of chondrocytes.60,61 Mitochondrial dysfunctions could break the balance between the glycolysis and OXPHOS, resulting in a significant reduction of ATP production in chondrocytes.
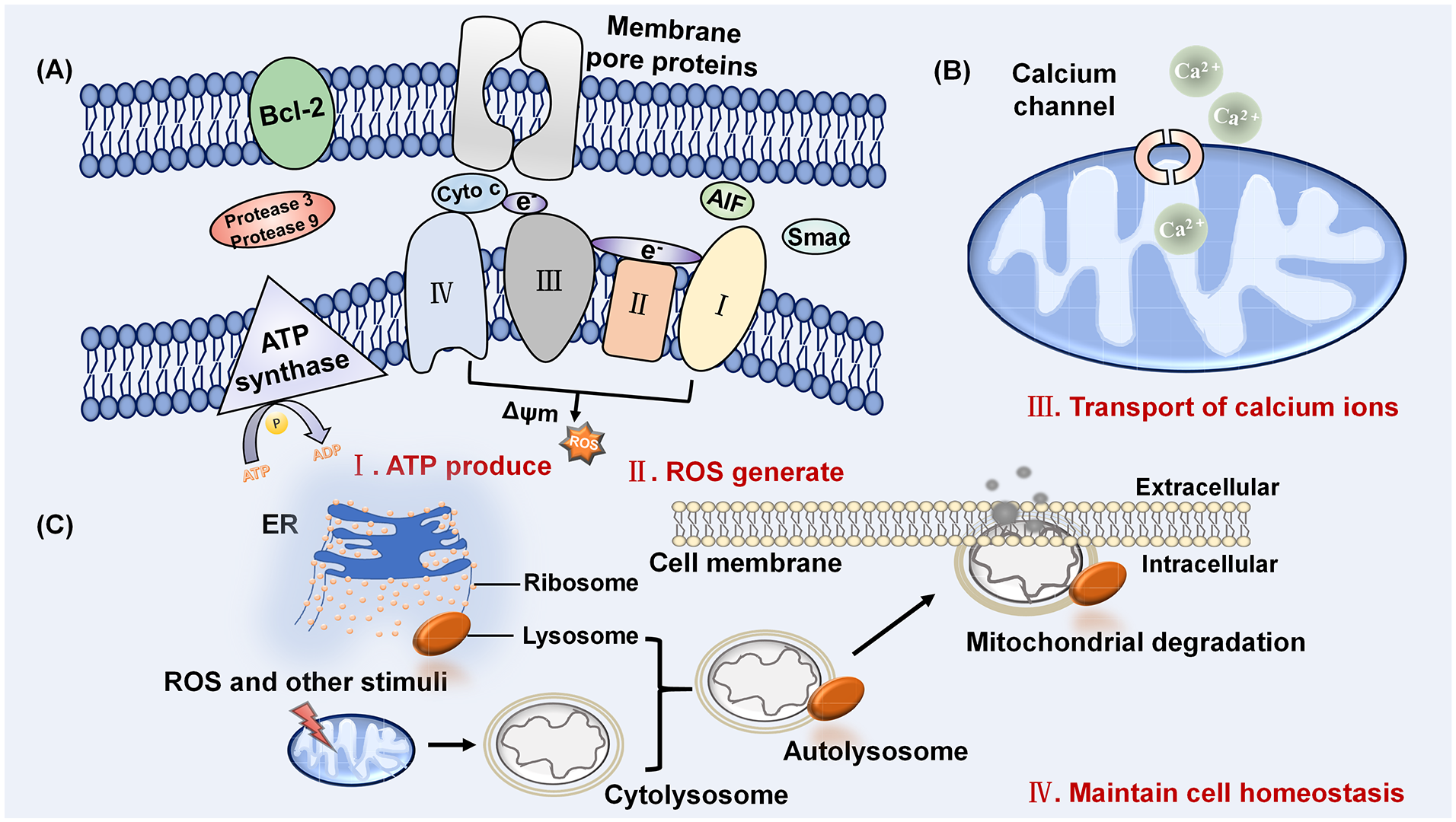
The function of mitochondria in chondrocytes. (
Chondrocytes are the only mature cell type in cartilage. Disorders of ATP synthesis in chondrocytes could also affect extracellular protein synthesis and matrix stability. 39 Glycolytic and TCA are upregulated to meet the demand of the ATP for cartilage maintenance and repair in metabolic diseases such as OA. Insufficient ATP production caused by mitochondrial dysfunction may disrupt the repair activities against cartilage degradation.62,63
Calcium Homeostasis
Calcium homeostasis is a basis of cellular metabolism. Several mitochondrial activities, such as ATP synthesis, are regulated by Ca2+ signaling. The high concentration of Ca2+ in mitochondria causes cell death, and the low concentration of calcium ions leads to disorders of cell energy metabolism.30,64 Normally, the endoplasmic reticulum (ER) releases Ca2+. Then, Ca2+ is transported into the mitochondrial matrix. In the mitochondrial matrix, the TCA cycle is activated by Ca2+ to stimulate ATP production ( Fig. 2B ). 65 Excessive Ca2+ leads to mitochondrial calcium overload, mitochondrial permeability transition pores (PTP) opening and chondrocyte apoptosis. 21 However, insufficient Ca2+ can cause ATP production impairments and metabolic disorders. Ca2+ stimulates mitochondrial enzymes to regulate the enzyme activity of the TCA cycle. Mitochondrial Ca2+ signaling dysregulation causes a decrease of ATP production and further affects ATP-related mitochondrial metabolism activity. 30 It seems that mechanotransductive calcium signals may also be related to mitochondrial polarization or depolarization, but the precise mechanism still needs to be explored. 66
Mitochondria play a role in regulating calcium homeostasis by a structure called mitochondria-associated endoplasmic reticulum membranes (MAMs). MAMs transport and regulate Ca2+ between the ER and mitochondria. 67 When calcium ions are released from the ER, the concentration of Ca2+ in some parts of mitochondrial surface is much higher than that in most areas of cytoplasm. As a calcium ion transfer bridge between the ER and mitochondria, MAMs provide a buffer area for Ca2+. 68 Inositol 1,4,5-triphosphate receptor (IP3R) is one of the most important calcium channels in the ER that controls the release of Ca2+. Voltage-dependent anion channel 1 (VDAC1) is a Ca2+-related protein located in the OMM that mediates mitochondrial uptake of Ca2+.69,70 The IP3R-VDAC1 complex is the most crucial structure associated with calcium ion transport in MAMs. 71 Once chondrocytes are stimulated, Ca2+ will be released from the ER by IP3R or ryanodine sensors (RyRs). These calcium ions then passively transported into mitochondria by high-conductance protein VDAC1. A high concentration of Ca2+ on the mitochondrial membranes activates the mitochondrial calcium uniporter (MCU) complex. 72 Driven by an electrical gradient, MCU mediates the transport of Ca2+ through the IMM. Subsequently, Ca2+ enters the mitochondrial matrix and participates in a series of metabolic reactions associated with energy production, cytosolic Ca2+ signaling, cell death, and calcification of the ECM. 73
Redox Regulation
Mitochondria are the places where oxygen free radicals are produced in chondrocytes. 46 Due to proton leakage in the ETC, mitochondria produce ROS, which are also called mtROS ( Fig. 2A ).74,75 The main sites of ROS production include incomplete OXPHOS in mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH), nitrogen oxidase (NOX), and xanthine oxidase (XO). 76 Mitochondrial electron transfer is generally considered to be a vital biological process that generates ROS under physiological conditions. The related enzymes in mitochondrial electron transfer including complex I, complex III, and glycerol-3-phosphate dehydrogenase (GHD). 77 In normal, an oxygen molecule receives 4 electrons transferred by the respiratory chain and is reduced in complex IX and interacts with 4 H+ to form 2 molecules of water. Once electrons reside in any unstable sites in the ETC, such as Fe-S clusters, flavin, or quinone, these sites are susceptible to single-electron oxidation by nearby oxygen. 78 During mitochondrial electron transport process, ETC directly transfers an electron to oxygen to generate superoxide anion (O2-) radical. A spontaneous or enzymatic mutation of O2- produces hydrogen peroxide (H2O2). And H2O2 further produces hydroxyl radical (∙OH). 79 As a participant in the intracellular signal transduction mechanism, ROS is an important cellular messenger in gene regulation including expression of matrix-degrading enzymes. ROS also contributes to maintaining chondrocyte homeostasis and regulates cell apoptosis, cytokine production, ECM synthesis, and decomposition.28,80
Mitochondria have a highly efficient antioxidant defense system to maintain a relatively low ROS level. In normal chondrocytes, only part of the ROS produced within mitochondria is released into the cytosol. ROS could also interact with a variety of mitochondrial components to reduce the escape of mtROS from mitochondria to the surrounding matrix. Superoxide removal system includes SOD, α-tocopherol or α-tocopheroxyl radical, cytochrome c, and some ions ligands such as Fe2+ and Fe3+. H2O2 is less reactive but important mediator of redox signaling. However, excessive production of H2O2 could also induce oxidative damage to DNA, lipids, and proteins. The scavenge of H2O2 involves GPX, which uses reduced glutathione as a substrate for the oxidative decomposition of H2O2. 77 Meanwhile, mitochondria could remove ROS that is produced by other cellular sources such as cytoplasmic enzymes and various oxidases. 79 Mitochondrial antioxidant system could also clear excessive O2∙- and H2O2 under some pathological conditions such as ischemia-reperfusion. 81
Furthermore, mitochondria are rich in DNA, proteins, and lipids, which are the target of mtROS attack. 82 The mtROS failed to be cleared increases the risk of damage to these mitochondrial components.83,84 Generally, excessive mtROS increases the risk of mtDNA mutations and ATP synthesis disorders and leads to mitochondrial dysfunction. 85 The theory of mitochondria and aging also shows that mitochondria are the main source and target of oxygen and nitrogen free radicals related to the aging process. The increase in ROS production leads to mtDNA damage. And the damage of mtDNA generates mutations of mitochondrial proteins and further increases the production of oxygen and nitrogen free radicals.2,86
Mitochondrial Autophagy
Mitochondria play a key role in the programmed apoptosis of dysfunctional cells. It is recognized that chondrocytes are difficult to repair. However, proper autophagy activation has a protective effect on OA chondrocytes and cartilage.87,88 Therefore, mitophagy plays a vital role in the survival of chondrocytes. 9 As the main site of aerobic respiration and energy supply in the cell, mitochondria are easily damaged. Damaged mitochondria are wrapped by a double-layer membrane structure and fuse with lysosomes to form autophagosomes. Then, enzymes degrade the contents of the autophagosome ( Fig. 2C ). 89 During the process of mitochondrial-mediated autophagy, the formation of mitochondrial autophagosomes is the most important step. 90 Through mitophagy, damaged or redundant mitochondria are promptly removed. This kind of cell-selective autophagy maintains the normal physiological function of cells as well as the stability of the intracellular space. 91
Mitophagy can be divided into 3 types according to the differences of mitochondrial autophagy receptors: receptors binding to ubiquitinated mitochondrial proteins, proteins localized in the OMM, and OMM-localized lipid molecules. 4 At present, the most studied mitochondrial autophagy pathways are the propeptide of phosphatase and tensin homolog–induced kinase 1 (PINK1)- and Parkin (PARK2)-mediated pathways. 92 In normal, PINK1 is cleaved by the mitochondrial processing peptidase (MPP) and releases to the cytoplasm. 93 PINK1 fragments directly bind to the E3 ubiquitin ligase Parkin, blocking its translocation and stabilization in the OMM, and act as mitophagy inhibitors. 94 The disruption of the ΔΨm causes the PINK1 to activate and localize in the OMM. Parkin is subsequently recruited and activated by PINK1. Parkin further selectively recruits autophagy receptors, such as nucleoporin 62/sequestosome 1 (p62/SQSTM1) and optineurin/optin, and initiates mitophagy. 95 Translocase of OMM 20 (TOMM20) is an OMM translocation enzyme, which acts as a marker of autophagy and degrades during autophagy. 96 Parkin and TOMM20 are positively and negatively correlated with mitophagy, respectively. 68 In addition to ubiquitin-binding receptors, OMM-localized proteins, such as Bcl-2/Adenovirus E1B 19 kDa-interacting protein 3 (Bnip3), Bnip3-like/NIP3-like protein X (Bnip3L/Nix), and FUN14 domain-containing protein 1 (FUNDC1), could also directly interact with lapidated LC3 (LC3-II), acting as mitochondrial autophagy receptors.97,98 Besides, in damaged mitochondria, OMM-localized lipids bind to lipidated form of LC3 (LC3-II) to induce the mitophagy. 99
Recently, mitochondrial Ca2+ has been verified to be a potentially specific signal that regulates mitophagy. The downregulation of mitochondrial Ca2+ could effectively reduce the activity of Parkin-mediated pathway. 100 And AMPK-dependent methods that enhance mitochondrial Ca2+ uptake could slow down the excessive activation of autophagic flux. 101 Hence, the regulation of mitophagy depends on the adjustment of mitochondrial Ca2+ to some extent.
However, mitophagy is a dual effect process. If oxidative stress damage exceeds the membrane potential of the mitochondrial inner membrane, dysfunctional mitochondria will be removed through the mitophagy. Although cells can remove damaged mitochondria and protect cells from apoptosis, excessive mitochondrial autophagy reduces a large number of important cell components and leads to cell death.102,103
Mitochondrial Biogenesis
Moreover, mitochondria are a kind of dynamic organelles. Mitochondria adjust their number, morphology, and position in cells by coordinating the cycle of mitochondrial fission and fusion. These changes regulate mitochondrial functions and cell metabolism. 104 Mitochondrial biogenesis controls the number, distribution, and morphology of mitochondria in the cell. The balance between fission and fusion is a key mechanism that regulates the structure and function of mitochondria and maintains the homeostasis of cells. 105 Some detailed studies have been performed on the molecular mechanisms that control the process of mitochondrial fission and fusion.37,106 In mammalian cells, mitochondrial fission is mainly driven by dynamin-related protein 1 (Drp1), a cytoplasmic dynamin guanosine triphosphatase (GTPase), dynamin2 (Dnm2), and mitochondrial fission protein 1 (Fis1). 107 The function of Drp1 is to participate in the fragmentation of mitochondria and peroxidase. Drp1 is dynamically recruited to OMM, that is, future fission sites, where it oligomerizes in a ring-like structure and drives membrane constriction in a GTP-dependent manner.108,109 Subsequently, Dnm2 is recruited to Drp1-mediated mitochondrial constriction neck that induces mitochondrial membrane scission. 110 Fis1 serves as a membrane-anchor that could also regulate mitochondrial fission through an interaction with Drp1, Dnm2 (and/or other fission components) to mitochondria.111,112 Mitochondrial fission contributes to the elimination of damaged mitochondria through mitophagy. In response to ATP demand, fission also benefits to the redistribution of mitochondria in chondrocytes. 113 However, mitochondrial fusion is controlled by 2 mitofusins (Mfn1 and Mfn2) and dominant optic atrophy 1 (Opa1). Mfn1 and Mfn2 are highly homologous, dynamically related GTPases with N-terminal GTPase domains. 114 Mfn1 and Mfn2 mediate the fusion of OMMs and then Opa1 mediates the fusion of IMM. 115 The outer membranes of 2 mitochondria are tethered by the trans-interaction of the HR2 and/or GTPase domains of Mfns. GTP binding and/or hydrolysis induces conformational change of Mfns, resulting in increased mitochondrial docking and membrane contact sites.116,117 Following OMM fusion, the interaction between OPA1 and cardiolipin (CL) on either side of the membrane tethers the 2 IMMs, which drive IMM fusion by OPA1-depedent GTP hydrolysis. 118 Mitochondrial fusion promotes the exchange of important components among mitochondria, especially mtDNA, and ensures the continuity of mitochondrial function. 119
Besides, mitochondrial mobility and morphology dynamics are related to cytoskeleton proteins, 120 cellular energy metabolism, and Ca2+ homeostasis. 121 And mitochondrial dynamics–related proteins are altered in many diseases, such as insulin resistanc, 122 type II diabetes, 123 neurodegenerative diseases, 124 Alzheimer’s disease, 125 cancer, 126 cardiovascular diseases, 127 and ischemic and dilated cardiomyopathy.128,129 Dynamic changes in mitochondria are critical for mtDNA integrity, energy generation, and cellular processes such as calcium signaling and apoptosis regulation. 130
Innate Immune Activities
Mitochondria have immune activities and play an important role in innate immune responses. Mitochondria are derived from α-bacteria that were engulfed by prototype cells. 131 During the process of cellular evolution, an endosymbiotic relationship gradually developed. Therefore, mitochondrial components have strong immune activity that is similar to bacteria. When the integrity of mitochondrial membrane is damaged, mitochondrial components, called damage-associated molecular patterns (DAMPs), are released to the cytoplasm. These endogenous molecules can be directly recognized by receptors to trigger immune responses. For instance, in the process of cell death or mitochondrial damage, the endogenously oxidized mtDNA released from the mitochondria is recognized as DAMPs, which trigger innate immune responses and cause inflammation. 132 Any mitochondrial event that results in mtDNA leakage could stimulate the cyclic guanosine monophosphate synthase stimulator of interferon genes (cGAS-STING) pathway and lead to the occurrence of innate inflammatory responses. 133 In addition to mtDNA, DAMPs also include structural phospholipid CL, n-formyl peptides (N-fp), and mtROS. 134 The process that eliciting an immune response has been verified to be associated with the NF-κB signaling pathway and induces the production of pro-inflammatory cytokines, including tumor necrosis factor-β (TNF-β). 135 In addition, several signal cascade steps in the innate immune response occur in the mitochondria or require the involvement of mitochondrial components. For example, mitochondrial component that play role in the formyl peptide receptors (FPRs) pathway is N-fp, which binds to FPRs and triggers neutrophil activation and migration. 136
Besides, metabolites of mitochondria and the metabolic states of mitochondria in cells could also regulate the immune response. The immune response triggers the release of cytokines and chemokines and recruits immune cells to repair mitochondrial metabolic damages. 137
Current Progress of Mitochondria in Chondrocyte Pathology
Once the morphology, structure, or function of mitochondria changed, chondrocytes will appear pathological state. Manifestations such as oxidative stress increase, chondrocyte apoptosis, inflammation-mediated matrix degradation, and cartilage matrix calcification were observed.60,138
Mitochondria show a regularly oval shape under normal physiological states. However, in a state of mitochondrial damage, irregularly shaped mitochondria increase and mitochondrial dense decreases. 38 Changes of mitochondrial structure such as the loss of folded cristae, the opening of mitochondrial PTP, and the damage of mitochondrial membrane suggest the pathological state of mitochondria. Due to the specific location and important role of mitochondria in ATP generation and cellular physiological activities, mitochondrial structure defects are related to mitochondrial-mediated diseases such as cartilage degradation diseases and Parkinson’s disease (PD).139,140 Mitochondrial dysfunctions are associated with impaired MRC, depolarized ΔΨm, increased mtROS, and dysregulated Ca2+.17,62 Studies have shown that mitochondrial dysfunctions are involved in the pathogenesis of OA, RA, and other cartilage-related diseases such as PTOA, Kashin-Beck disease (KBD), cartilage degeneration, and cartilage-mediated growth retardation.141-145
However, whether these mechanisms in mitochondrial morphology, structure, and function directly are directly related to chondrocyte abnormalities still need to be studied. How mitochondrial abnormalities lead to the occurrence of OA disease is still a new field in the future.
The Mitochondria in OA Pathogenesis
OA is a degenerative rheumatic disease associated with the degradation of articular chondrocyte-derived ECM and chondrocyte apoptosis. The main pathological features of OA are osteophyte formation, subchondral bone sclerosis, abnormal blood vessels, and articular cartilage degeneration. Patients with OA usually exhibit chronic pain and articular dysfunction. 146 The role of mitochondria in degenerative diseases is widely recognized. 147 In 2002, Terkeltaub et al. 39 published a review that first described chondrocyte mitochondrial damage as an important mediator of OA. At present, several studies have shown that mtDNA haplogroups are related to OA and cluster TJ have a protective role in the prevalence of OA risk. 148 A research reported that mtDNA haplogroup type B reduces the risk of OA, whereas type G increases the risk in a southern Chinese population. 149 However, these results are not universal among Asian population up to now. 150
Mitochondria have a profound impact on the pathogenesis of OA, including the inhibition of respiratory chain activity, the increased generation of ROS, the disbalance of calcium homeostasis, the activation the mitochondrial-mediated apoptosis, and the generation of inflammation in chondrocytes and cartilage ( Fig. 3 ).
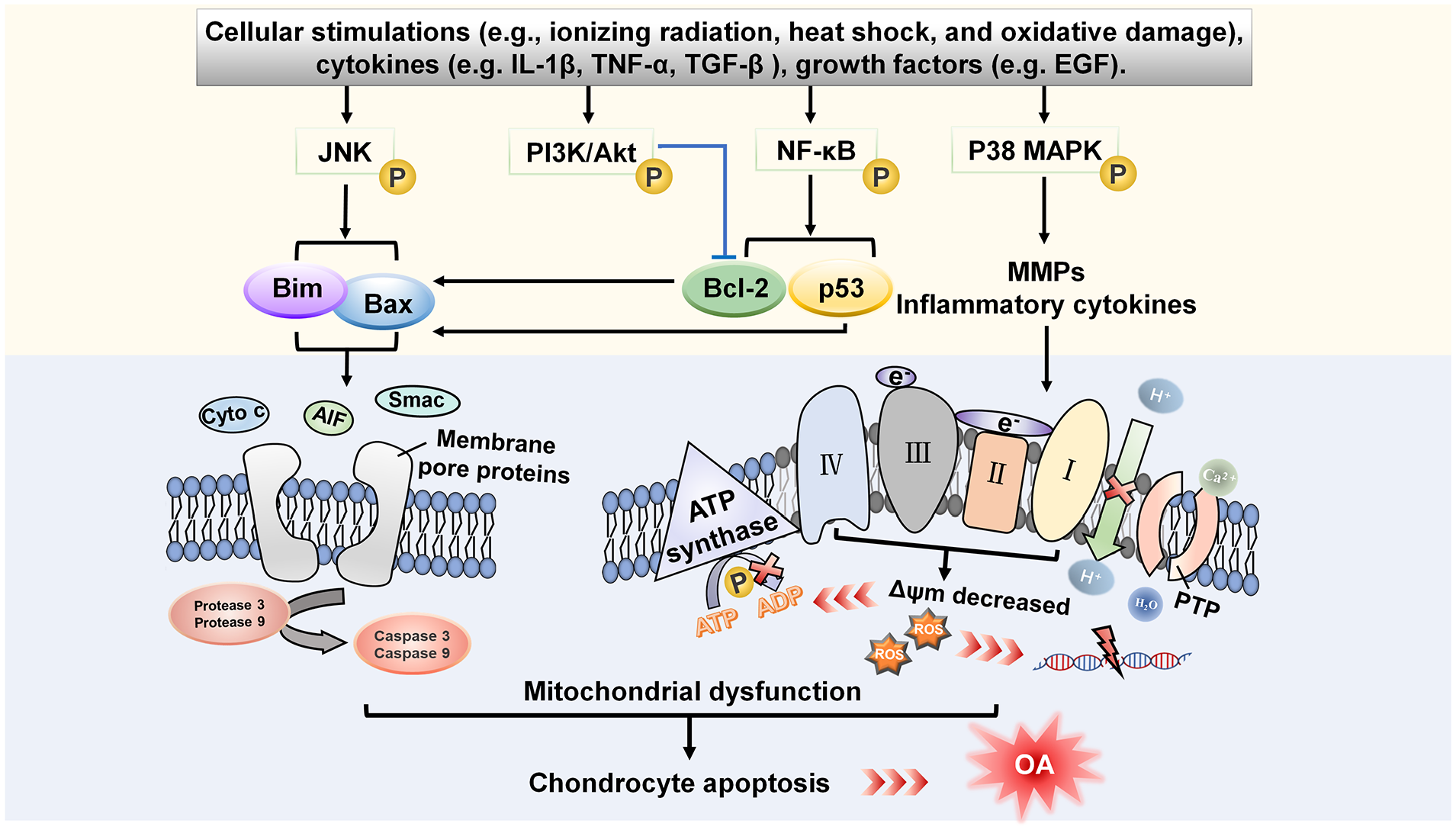
Schematic diagram of mitochondrial function of chondrocyte in OA. Cytokines and other stimuli mainly lead to chondrocytes apoptosis through mitochondrial apoptosis pathway. The key chondrocyte signal transduction pathways include the JNK pathway, the PI-3K/Akt pathway, the NF-κB pathway, and the p38/MAPK pathway. Phosphorylation and inactivation of the anti-apoptotic protein Bcl-2 could suppress its anti-apoptotic activity and induce the escape of Cyto c. The JNK and NF-κB pathways promote p53 to upregulate pro-apoptotic genes such as Bax and Bim. The aggregations of these pro-apoptotic molecules and pro-inflammatory cytokines further impair mitochondrial integrity. Compared with normal chondrocytes, the MRC of OA chondrocytes showed that the activity of complexes I, II, and III and ΔΨm were significantly decreased. Inhibition of MRC can lead to insufficient electron transport, causing a decrease in ATP production. Mitochondrial damage not only reduces the efficiency of bioenergy production, but also increases the ROS production. Antioxidant defense system cannot eliminate excessive ROS and lead to oxidative stress. Then, oxidative stress interferes with cell regulation mechanism and further causes cell apoptosis. Excessive mtROS also increases the risk of mtDNA mutations and ATP synthesis disorders, which deteriorating mitochondrial dysfunction. At the same time, mitochondrial dysfunction disrupts Ca2+ homeostasis. Excessive accumulation of Ca2+ leads to the opening of mitochondrial PTP and the damage of mitochondrial membrane structure. The entry of H2O molecules into the mitochondria leads to an increase in mitochondrial mass, which ultimately results in the swelling and the degradation of mitochondria. In damaged mitochondria, pro-apoptotic cytokines such as Cyto c escape from the mitochondrial intermembrane space via membrane pore proteins and bind to cytoplasmic scaffolding protein Apaf-1 to activate protease 9 and 3. Pro-apoptotic proteins induce the occurrence of the mitochondrial-mediated apoptosis pathway. Finally, chondrocyte apoptosis promotes the occurrence of OA. OA = osteoarthritis; JNK = c-Jun N-terminal kinase; PI-3K/Akt = phosphatidylinositol-3-kinase-protein kinase B; NF-κB = nuclear factor-κB; MAPK = mitogen-activated protein kinase; Bcl-2 = B-cell leukemia/lymphoma-2; Bax = Bcl-2-associated X protein; Bim = Bcl-2 interacting mediator of cell death; MRC = mitochondrial respiratory chain; ΔΨm = mitochondrial membrane potential; ATP = adenosine triphosphate; ROS = reactive oxygen species; mtROS = mitochondrial reactive oxygen species; mtDNA = mitochondrial deoxyribonucleic acid; PTP = permeability transition pores; Cyto c = cytochrome c; Apaf-1 = apoptotic protease activating factor-1; AIF = apoptosis-inducing factor; Smac = second mitochondrial-derived activator of caspases; MMPs = matrix metalloproteinases.
The destruction of the MRC could cause OA. The MRC analysis of OA chondrocytes showed that the activity of complexes I, II, and III and the ΔΨm were significantly decreased. 151 Compared with normal chondrocytes, Maneiro et al. 58 found that the activity of MRC complexes II and III in patients with OA decreased by 35% and 13%, respectively, and the diminished efficiency of transporting electrons cannot be compensated by the increased activity of complex I. These attenuations lead to a blockade of cartilage synthesis, chondrocyte apoptosis, cartilage matrix calcification, and inflammatory responses. 62 The dysfunction of complexes II and III leads to an increase in mitochondrial mass, which is a compensatory response to the decrease of ATP production caused by inadequate electron transport. 58 And the change of citrate synthase (CS) activity may be an indicator of mitochondrial mass.140,152 Cytokines, such as TNF-α and IL-1β, can play a role in cartilage degradation by reducing MRC complex I activity, ATP production, and mitochondrial membrane potential. 51 Studies have reported that the inhibition of MRC enhanced the expression of COX-2 and PGE2 in normal human chondrocytes 53 as well as the inflammatory responsiveness to cytokines. 153
Damaged mitochondria reduce the efficiency of bioenergy production and increase the production of ROS. Subsequently, the increase of ROS interferes with cell control mechanisms and leads to cell apoptosis. 154 In a pathological state, antioxidant defense system cannot remove excessive ROS and result in oxidative stress, even apoptosis. 28 In general, excess mtROS increases the risk of mtDNA mutations and ATP synthesis disorders. Ultimately, mtROS that cannot be cleared causes mitochondrial dysfunction. 85 Studies have shown that mtDNA damage and respiratory failure give rise to elevated intracellular oxygen tension and unstable hypoxia-inducible factor-1α (HIF-1α) level, resulting in cell death.33,155 Hypoxic environment is a highly sensitive environment for cells. Mitochondrial dysfunction in hypoxia could cause the depolarization of ΔΨm, which promotes ROS production via NADPH oxidase. 156 In addition to hypoxia environment, age-related oxidative stress could also lead to the increase of ROS level. 157 Oxidative stress in human chondrocytes accelerates senescence and telomere shortening. 158 Excessive ROS, as a second messenger, activates related signaling pathways such as PI-3K-Akt and promotes cartilage degradation by inhibiting matrix synthesis, cell migration, and growth factor biological activity.25-27 Moreover, the overproduction of ROS also affects mitochondria function and causes mtDNA damage. And then, mitochondrial dysfunction together with mtDNA damage contributes to cartilage degeneration. 159 ROS can also enhance the inflammatory response caused by the increase of chemotactic and pro-inflammatory cytokines. The exacerbation of the inflammatory response leads to mtDNA damage, lipid peroxidation, collagen scission, respiratory chain inhibition, and ATP synthesis decrease. The damage to the MRC further aggravates the pathological changes of mitochondria, forming a vicious circle.7,160
Abnormal mitochondrial function is related to calcium ion homeostasis disorder. Excessive accumulation of Ca2+ leads to mitochondrial PTP opening and mitochondrial depolarization. These mitochondrial changes eventually result in mitochondrial swelling and cell apoptosis. Hypoxia in articular cartilage promotes an increase in intracellular Ca2+ levels (also called cytoplasmic Ca2+), which is associated with mitochondrial depolarization and a decrease in ROS levels. 161 Moreover, due to the inhibition of MRC complexes III and V activity, the expression of interleukin-8 (IL-8), cyclooxygenase-2 (Cox-2), and prostaglandin E2 (PGE2) increased. The overexpression of these cytokines promotes mitochondrial Ca2+ overload. Then, excessive Ca2+ activates the NF-κB signaling pathway and upregulates MMP-1, MMP-3, and MMP-13 expression to promote cartilage degradation. 161 Meanwhile, the suppression of mitochondrial respiration promotes MVs-mediated mineralization in chondrocytes and the deposition of pathologic calcium pyrophosphate dihydrate (CPPD) crystal as well as hydroxyapatite (HA) crystal. 60
Cartilage diseases are closely related to abnormal mitochondrial-mediated apoptosis. Previous studies have also shown that mitochondrial apoptosis is an important pathological process in OA.42,153 Chondrocyte apoptosis exhibits a special characteristic that corresponds to the unique type of programmed cell death called cartilage ptosis. 22 Mitochondrial changes are one of the most typical manifestations in cell death,162,163 such as the loss of MMP and the decline in ATP production.143-145,164 In the process of chondrocyte apoptosis, the increase of membrane permeability and the release of proapoptotic factors such as Cyto c are also related to mitochondria. 165
There is an interaction between pro-inflammatory cytokines and mitochondria that pro-inflammatory cytokines also play a role in the dysfunction of mitochondria. Studies have shown mitochondrial dysfunction increased the expression of pro-inflammatory cytokines, such as IL-1β, TNF-α, and nitric oxide (NO) in chondrocytes. 153 The responses to pro-inflammatory cytokines mainly involve the enhancement of superoxide in chondrocytes and subsequently activate redox-sensitive transcription factor NF-κB. 166 Then, the mitochondria break down and activate the apoptosis pathway, clearing the damaged mitochondria. 167 Researches have confirmed that oxidative stress induced by IL-1β treatment in OA chondrocytes is associated with increased apoptosis. Ansari et al. 168 found that Butein could activate AMPKα/TSC2/ULK1/mTOR pathway under pathological conditions to activate autophagy which is essential for inhibiting the expression of inflammatory mediators in OA chondrocytes. However, the mitochondrial dysfunction of chondrocytes may increase the inflammatory responses induced by cytokines. Pro-inflammatory cytokines contribute significantly to the initiation and progression of OA via upregulation of catabolic process. Inflammatory responses further aggravate the mitochondrial pathology and form a vicious cycle. Pro-inflammatory cytokines lead to mtDNA damage and decreased mitochondrial transcription. These injuries are also associated with the increase of NO and chondrocyte apoptosis. 169 Hence, inducible NO synthase (iNOS) inhibitors showed a chondroprotective effect and it may be at least partly due to the reduction in IL-1β-induced mitochondrial dysfunction. 170
The excessive degradation of cartilage matrix is considered to be mainly caused by the high expression of inflammatory factors. Studies have suggested that the degradation of cartilage matrix leads to pathological changes of articular cartilage, which may be the result of the effects of MMPs, ADAMTS, NO, substance P (SP), and PGE2.171,172 NF-κB and MAPK signaling pathways are activated by IL-1β and affect gene transcriptional modification in the nucleus. Among these inflammatory factors, the level of IL-1β and TNF-α in OA patients increased significantly.173,174 IL-1β and TNF-α have a similar mechanism that could change chondrocyte mechanical properties by altering F-actin cytoskeleton. 175 IL-6 was also involved in both inflammatory immune cartilage destruction and subchondral bone reconstruction in OA. 176 The combined function of these 3 factors can inhibit the production of PGs, increase the synthesis of MMPs, and aggravate cartilage loss. MMPs can cause the denaturation of type II collagen and increase the loss of PG, which is an important enzyme that regulates the degradation of cartilage matrix. MMPs can be divided into more than 10 subtypes. MMP-3 can degrade most collagen. The degradation ability of MMP-9 and MMP-13 on cartilage matrix can also be activated and amplified by MMP-3. Both MMP-3 and MMP-13 can directly degrade type II collagen, aggravating the damage of cartilage. 177 To maintain the normal structure and function of cartilage, the activity of MMPs can be inhibited by tissue inhibitors of metalloproteinases (TIMPs). MMPs and TIMPs are relatively balanced in normal. 178
In brief, chondrocyte mitochondrial dysfunction, such as MRC inhibition and oxidative stress, is thought to be involved in the mechanisms of OA. A mitochondrial proteomics study of OA chondrocytes confirmed changes in mitochondrial protein expression significantly changed proteins including energy production, maintenance of mitochondrial membrane integrity, oxidative stress response, and apoptosis-associated proteins. 179 Mitochondrial dysfunction may disrupt the repair activities against cartilage degradation and promote the production of oxidative stress and inflammatory mediators. These pathological changes further cause chondrocyte apoptosis and inflammation, thereby promoting the occurrence of OA.
The Pathological Role of Mitochondria in RA
RA is a multifactorial systemic autoimmune disease. RA is characterized by synovial inflammation and hyperplasia, autoantibody production, cartilage and bone destruction, and systemic manifestations including cardiovascular, lung, and bone diseases. The main pathological features of RA are chronic synovitis with hyperplasia, pannus formation, and immune cell infiltration. 180 The pathological role of mitochondria in RA is apparently critical ( Fig. 4 ).
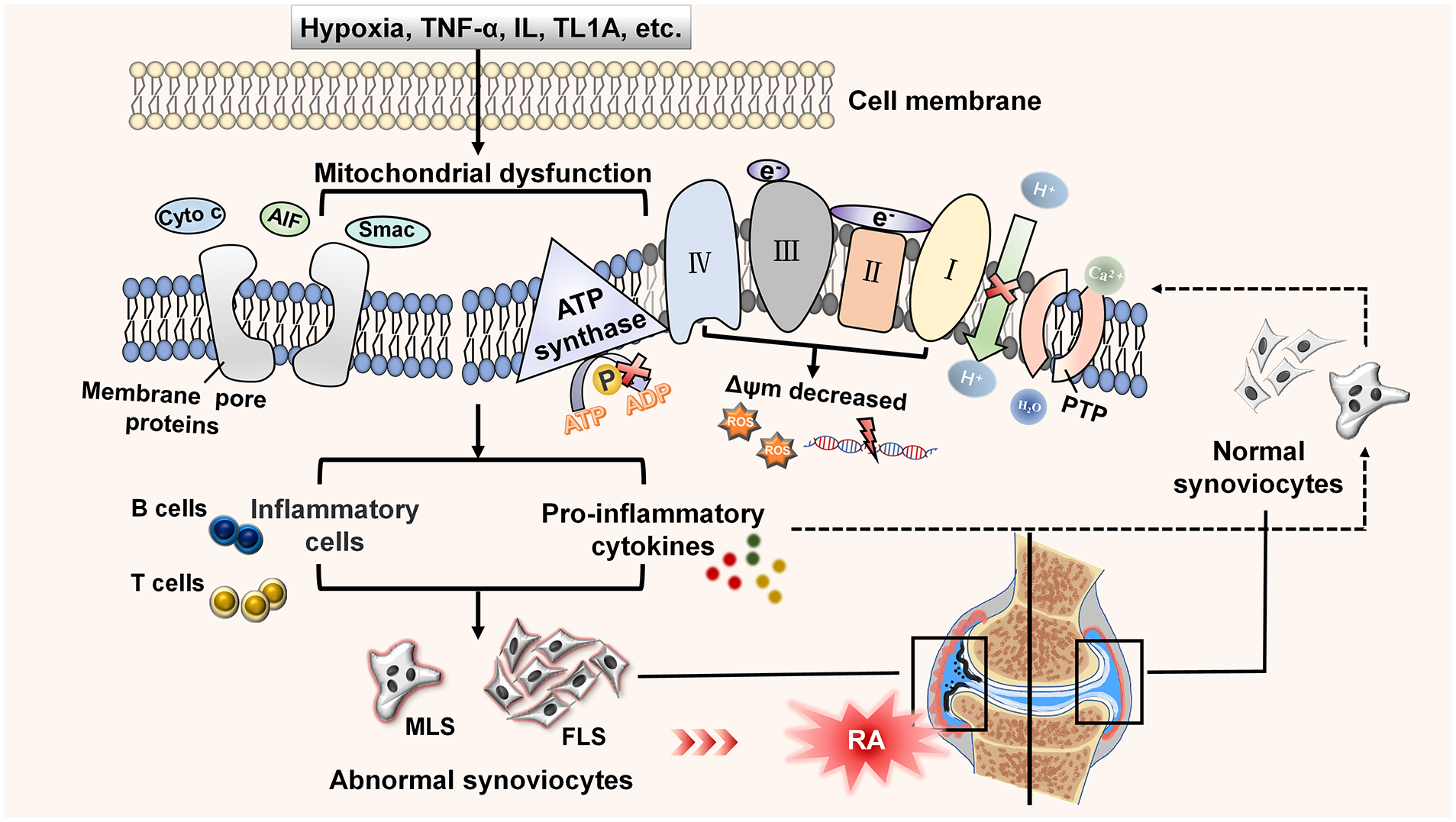
Schematic diagram of mitochondrial function in RA. In RA, the microvascular disorders of synovial tissue cause hypoxia. Hypoxia induces mitochondrial dysfunction that promotes the release of ROS. The accumulation of ROS promotes the occurrence of chondrocyte apoptosis. At the same time, the aggregation of pro-inflammatory mediators and inflammatory cells induces the inflammatory response of synovial cells. These inflammatory mediators released by mitochondrial dysfunction stimulate normal cells to cause mitochondrial damage, forming a vicious cycle. Abnormal mitochondrial structure and function also inhibit the apoptosis of synoviocytes and promote cells proliferation. RA = rheumatoid arthritis; ROS = reactive oxygen species; TNF-α = tumor necrosis factor-α; IL = interleukin; Cyto c = cytochrome c; AIF = apoptosis-inducing factor; Smac = second mitochondrial-derived activator of caspases; ATP = adenosine triphosphate; PTP = permeability transition pores; MLS = macrophage-like synoviocytes; FLS = fibroblast-like synoviocytes.
Previous reports suggested that ROS is the basis for these RA pathological characteristics. And oxidative stress is thought to be associated with the onset of RA. 181 The production of ROS is closely related to mitochondria. Therefore, mitochondria are considered to be important organelles in the pathogenesis of RA. Fluctuations of ROS levels are associated with mitochondrial dysfunction, especially under hypoxia conditions. In RA, the microvascular disturbance of synovial tissue can lead to the generation of anoxic environment. The hyperplasia of pannus leads to an increase of energy demand for mitochondrial electron transport. Hypoxia and ATP demand increase mtROS production and further promote glycolysis and inflammation responses.8,182 The pro-inflammatory activities are activated by HIF-1α, NF-κB, Janus kinase-signal transducer and activator of transcription (JAK-STAT), activator protein 1 (AP-1), and notch pathways. 183 ROS-mediated mitochondrial damage may also lead to cartilage destruction by the upregulation of MMPs. 28 ROS can also directly or indirectly damage cellular components, such as the ECM, by inducing matrix degradation mediators.184,185 At the site of cartilage injury, ROS can hinder the effect of growth factors on chondrocytes, especially the combination of chondrocytes and ECM, and ultimately lead to chondrocyte apoptosis.185,186
The genetic factors of mitochondria also play a role in the pathogenesis of RA. Studies have shown that there is a significant correlation between severe RA and MRC-related genes. 187 MRC is the main site of ROS production, which also suggests that there is a correlation between RA and MRC dysfunction. A study investigated the function of 1,158 nuclear DNA (nDNA)-encoded proteins that translocate to mitochondria. Results showed that the activity of oxidation-reduction enzymes was affected in RA. And the change of enzymes activity is related to the destruction of mitochondrial oxidative phosphorylation complexe. 183 The dysfunction of mitochondrial oxidation-reduction enzyme causes oxidative stress, such as the activation of ROS. ROS changes the metabolism of chondrocytes and upregulates the level of MMPs. Finally, the dysfunction of mitochondria results in the degradation of cartilage and the development of inflammation. 74 RA-related single nucleotide variations (SNVs) are associated with excessive ROS that are induced by increased electron leakage in MRC. 187 The accumulation of rare/low-frequency variations in the MRC suggests a genetic basis of oxidative stress in RA patients.
Chondrocyte autophagy may also be another etiology of RA. However, there are few reports on the autophagy of RA chondrocytes. The pathophysiological mechanisms of cartilage degradation in RA and OA are similar to some extent. Therefore, the chondrocyte autophagy of RA and OA may also have a similar mechanism. 36 Mitochondrial dysfunction is characterized by ΔΨm depolarization and usually accompanied by the release of mitochondrial proapoptotic proteins, such as Cyto c, AIF, and Smac. When the stress on the cells is severe or prolonged, the calcium balance is broken. ER moves Ca2+ to mitochondria, which impairs the function of mitochondrial and triggers the proapoptotic signals of chondrocytes. In addition to Ca2+ imbalance, oxidative stress also accelerates apoptosis by advanced oxidation production products (AOPPs).188,189 And AOPPs are products containing dityrosine and cross-linking proteins that accumulate in RA patients. The exposure of chondrocytes to AOPPs may lead to apoptosis. 190 AOPPs stimulate the generation of ROS and lead to mitochondrial dysfunction. Then, the caspase family is activated by mitochondrial dysfunction, inducing chondrocyte apoptosis through the endogenous apoptotic pathway. 191
The association between mitochondrial dysfunction and inflammation in RA has also been established. Damaged mitochondria could trigger immune and pro-inflammation responses. Oxidized mitochondrial components such as mtDNA, proteins, and lipids accumulate in inflamed rheumatoid joints. The accumulation of these oxidized components promotes the production of autoantibodies and drives the occurrence of autoimmune response. 192 Immune responses promote the release of inflammatory mediators and the accumulation of inflammatory cells, thereby increasing oxidative stress. Oxidative stress leads to the abnormalities in normal mitochondria and forms a vicious cycle.133,193 Mitochondrial dysfunction induces low-grade inflammatory response in RA-related cells, such as synoviocytes, and increases cell sensitivity and the expression of cytokine-induced inflammatory mediators. The occurrence of this process depends on the generation of ROS and the activation of NF-κB. 194
The inflammatory microenvironment in OA caused by synoviocyte resistance is also associated with the occurrence of RA. Disorders of mitochondrial-related apoptosis pathways affect the apoptosis of synovial cell. Whereafter, synoviocyte resistance occurs.195,196 The resistance of synoviocyte to apoptosis is a multifactorial process. However, it is certain that mitochondrial changes play an important role in synoviocyte resistance. 197 Studies have shown that hypoxia-induced mitochondrial abnormalities in rheumatoid arthritis fibroblast-like synoviocytes (RAFLSs) may induce the secretion of basic fibroblast growth factor (Bfgf) in vitro and lead to cell proliferation. During the development of synovial cell inflammation, the anti-apoptotic factor Bcl-2 increased, and the apoptotic factor caspase 8 and Cyto c decreased. This may also be the reason why synovial cells resist apoptosis. In addition, studies have shown that mtROS could increase the burden of mtDNA mutation. If the genes that promote cell survival are mutated, mtDNA mutations could promote the proliferation of synovial cells. 31 Fibroblast-like synoviocytes (FLSs) produce a variety of enzymes, such as MMPs and collagenase, which are of great significance for enhancing cell invasiveness and tissue destruction. In addition, the invading synovial cells express a variety of adhesion molecules, such as vascular cell adhesion protein 1 (VCAM-1) and several integrins. These adhesion molecules induce adjacent chondrocytes to produce ECM degradation enzymes and lead to the decomposition of ECM and the onset of RA. 198
Conclusion and Perspectives
Mitochondria are indispensable energy-producing organelles in chondrocytes. In addition to producing ATP, mitochondria also participate in many key physiological processes and play an important role in cellular activities, including programmed cell death, calcium homeostasis, tissue damage, innate immune response, intermediate metabolites oxidation, and cell homeostasis regulation.134,199,200 The integrity of mitochondrial morphology, structure, and function is essential for maintaining the normal physiological states of cartilage and chondrocytes. Mitochondrial dysfunction plays a key role in the pathogenesis of cartilage-related diseases such as OA ( Table 1 ) and RA ( Table 2 ). Dysfunction of mitochondria in chondrocytes induces and exacerbates inflammation, promotes cell apoptosis, and causes cartilage degradation. However, changes in the levels of various factors and signaling pathways in cartilage and chondrocytes can also lead to the abnormalities of mitochondrial structure and function. Chondrocytes are important for the degradation of cartilage in response to inflammatory stimuli. However, this does not mean that chondrocytes are the only participants in joint inflammation. Studies have also shown that other cells such as synovial cells and macrophages are involved in the process of joint inflammation. 29
The Role of Mitochondria in OA.
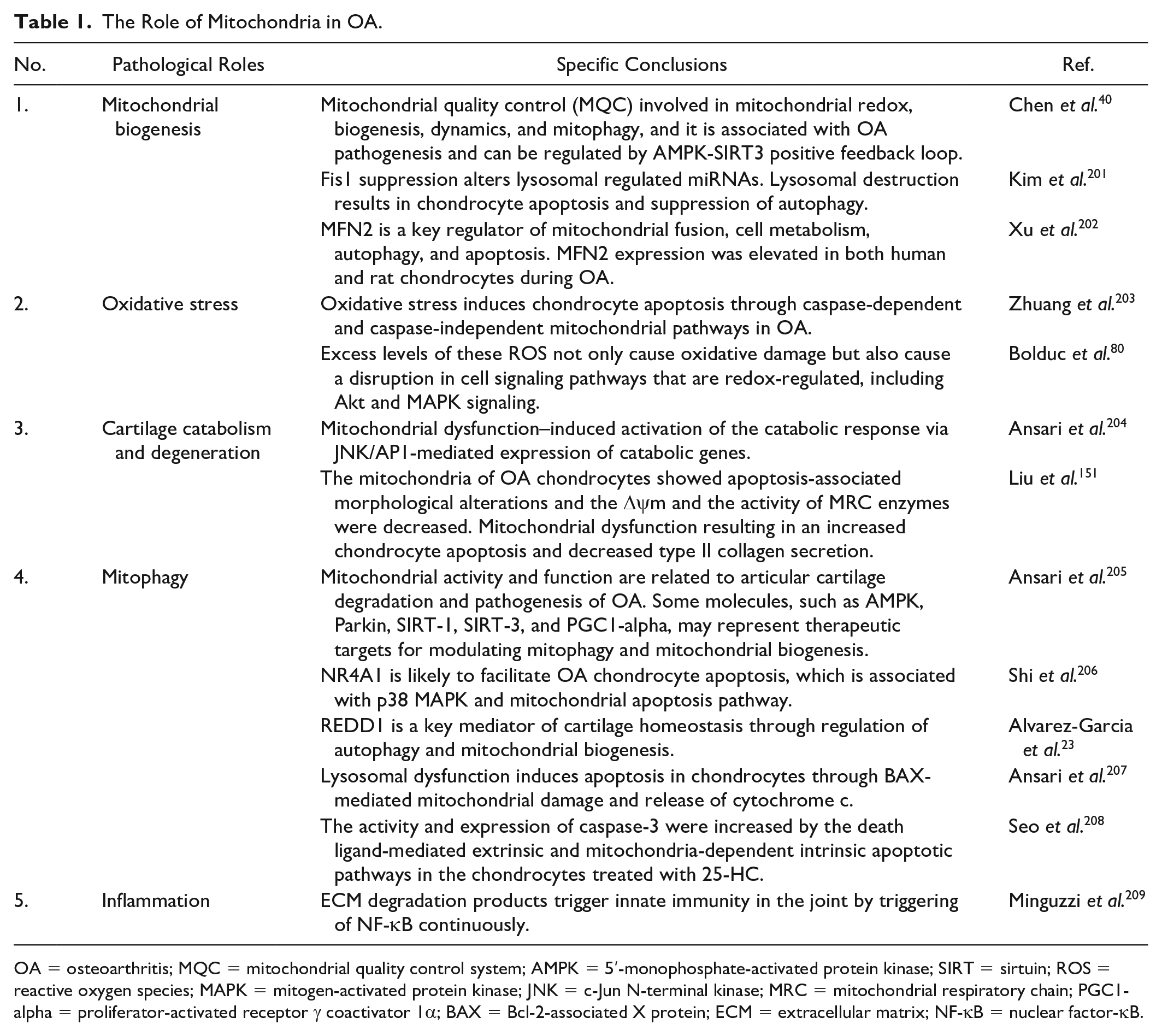
OA = osteoarthritis; MQC = mitochondrial quality control system; AMPK = 5′-monophosphate-activated protein kinase; SIRT = sirtuin; ROS = reactive oxygen species; MAPK = mitogen-activated protein kinase; JNK = c-Jun N-terminal kinase; MRC = mitochondrial respiratory chain; PGC1-alpha = proliferator-activated receptor γ coactivator 1α; BAX = Bcl-2-associated X protein; ECM = extracellular matrix; NF-κB = nuclear factor-κB.
The Research Progress of Mitochondria in RA.

RA = rheumatoid arthritis; AOPP = advanced oxidation production product; ROS = reactive oxygen species; ER = endoplasmic reticulum; NO = nitric oxide; MRC = mitochondrial respiratory chain.
There is no doubt that mitochondria play an important role in cartilage-related diseases. However, whether changes in mitochondrial function and structure directly induce chondrocyte-related diseases remains to be explored. Although many researchers have confirmed that mitochondria play essential roles in cartilage and chondrocytes, studies on the specific mechanism are still limited. The mechanism of mitochondria inducing apoptosis of chondrocytes or other types of cells needs to be further studied. In the development of cartilage diseases, how to avoid the adverse effects of oxidative damage, inflammation, calcium homeostasis imbalance, and autophagy caused by mitochondria requires further research.
Footnotes
Acknowledgments and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (grant numbers 81600840 and 81771047 to Jing Xie, 11902058 to Chunli Wang).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.