
Abstract
The nonreceptor tyrosine kinases Abl and Arg are among the most well-characterized tyrosine kinases in the human genome. The activation of Abl by N-terminal fusions with Bcr (Bcr-Abl) or Gag (v-Abl) is responsible for chronic myeloid leukemia or Ph+ acute lymphoblastic leukemia and mouse leukemia virus, respectively. In addition, aberrant Abl and Arg activation downstream of several oncogenic growth factor receptors contributes to the development and progression of a variety of human cancers, often associated with poor clinical outcome, drug resistance, and tumor invasion and metastasis. Abl activation can occur by a variety of mechanisms that include domain interactions involving structural remodeling of autoinhibited conformations as well as direct phosphorylation by upstream kinases and phosphatases. Constitutive activation of Abl plays a significant role in regulating the actin cytoskeleton by modulating cell adhesion, motility, and invadopodia. This review addresses the role of Abl and Arg in tumor progression with particular emphasis on the roles of Crk and Abi1 adapter proteins as distinct molecular switches for Abl transactivation. These insights, combined with new insights into the structure of these kinases, provide the rationale to envision that Crk and Abi1 fine-tune Abl regulation to control signaling to the cytoskeleton.
Introduction
Since the discovery of adaptor proteins more than 20 years ago, there have been remarkable advances in the field of signal transduction, most notably from the realization that signaling proteins possess protein-protein and protein-lipid interacting domains that permit the assembly of large multiprotein complexes.1-3 Indeed, the “protein-protein interactome” has taken center stage in signal transduction, with the daunting challenges now to understand how complex signaling assemblages are regulated in time and space.4,5 Many of these assemblages are further regulated by protein posttranslational interactions, most notably by phosphorylation, adding complexity with respect to the regulation of interactomes but also clinical relevance in cancer as both the level of tyrosine phosphorylation and the expression of adaptor proteins are dysregulated in cancer.6,7 In this review, we discuss how the Crk and Abi1 adaptor proteins interact with Abl and propose a binary mechanism for Abl regulation under both physiological conditions and during cancer progression.
Abl and Arg Kinases
The nonreceptor tyrosine kinases, Abl and Abl-related gene (Arg), control several physiological processes for both development and tissue homeostasis.8,9 Homozygous deletions of both Abl and Arg (Abl/Arg DKO) die embryonically at late gestation accompanied by cardiovascular lesions and hemorrhages, and neurulation defects both linked to alterations in the actin cytoskeleton. 10 The knockout of Abl in an Arg-sufficient background has a milder phenotype than the Abl/Arg DKO but displays defects associated with deficient T-cell receptor signaling, manifesting thymic atrophy, leukopenia, and sensitivity to infections.11,12 In addition to controlling actin-dependent morphogenetic programs during embryogenesis and physiological responses in somatic cells, Abl is important for the pathogenesis and virulence determinants of Helicobacter pylori13,14 and Shigella flexneri.15,16 On the other hand, gain-of-function–activated species of Abl and Arg are implicated in tumor invasion and progression in a variety of cancers, particularly to the progression to metastatic disease, making them attractive targets for selective anticancer therapies.
Oncogenic Roles for Abl in Leukemia and Solid Cancer
Abl was first recognized as an oncogene encoded from the acutely transforming Abelson murine leukemia virus (v-Abl or Gag-Abl) that could directly transform both hematopoietic cells and NIH3T3 cells in culture. 17 Animals infected with Mo-MuLV developed multiple tumors of the lymph nodes with bone marrow infiltration. 18 The disease phenotype and progression of MuLV are reminiscent, but not identical, of the disease phenotype in chronic myeloid leukemia (CML) and Ph+ acute lymphoblastic leukemia (ALL), which is mediated by an independent N-terminal fusion of Bcr to Abl. 19
In contrast to CML and Ph+ ALL where Abl is a driver mutation, in solid cancers such as melanoma,20,21 non–small cell lung cancer, 22 colorectal cancer, 23 and breast cancer,24-26 Abl is activated downstream of activated growth factor receptors, as outlined by Plattner in this monograph. The mechanisms by which growth factor receptors activate Abl are multifactorial. In the case of PDGFRα and ErbB2, Abl can bind directly to the phosphorylated receptors via the Abl SH2 domain, causing SH2 domain replacement, that results in phosphorylation of the activation loop of the Abl kinase domain (see below). 27 Abl can reciprocally phosphorylate receptor tyrosine kinases in their cytoplasmic tails, leading to reinforced downstream signaling from pre-existing activated RTKs.28,29 A second and perhaps more physiological manner for growth factor–induced activation of Abl occurs indirectly and requires at least 2 types of receptor-activated signaling molecules. First, Src kinases that directly associate with PDGFR and become activated 30 , phosphorylate the activation loop of Abl and at a second site in the linker between the SH2 domain and the kinase domain to activate Abl. Second, Abl activation requires PLC-γ–dependent hydrolysis of PIP2; the latter serves as an inhibitor for Abl.31,32 The importance of Abl in PDGFa signaling is highlighted in that PDGF-inducible dorsal ruffling is defective in Abl/Arg DKO fibroblasts and can be rescued by re-expression of either Abl or Arg.27,33,34 This latter model is also reminiscent of the model of Abl activation downstream of TCR signaling in lymphocytes, where Abl activation requires Lck and its substrate ZAP70.35,36 While the necessity of Src for RTK-mediated Abl activation diversifies the signaling paradigm of RTK activation, the contribution of direct activation and indirect activation is not fully understood.
Dual Structures of Abl and Complex Interplay between Closed and Open Configurations
Although the regulation of Abl is clearly complex, X-ray crystallography models elegantly show that under physiological conditions, Abl activity is negatively regulated by multiple structural-dependent autoinhibitory mechanisms. In fact, autoinhibition has emerged as the mechanism of regulation for most, if not all, nonreceptor tyrosine kinases and particularly well described for c-Src and c-Abl, which are noted at the level of X-ray crystallography analysis37,38 and confirmed by rationale mutagenesis that unlocks key regulatory elements (see Hantschel in this issue). Nonreceptor tyrosine kinases share considerable structural homology conferred by the presence of the highly conserved structural domains of SH3 and SH2, which are positioned to tether against the catalytic kinase domain. Such 3-dimensional structures of c-Src39,40 and c-Abl41,42 revealed that the SH3 and SH2 domains bind to the catalytic domain, inducing an autoinhibitory conformation, which provides the basic mechanism of regulation of these kinases.
While both Src and Abl kinases are controlled by autoinhibition, they in fact differ from each other in their inhibitory mechanisms. For c-Src, inhibition is achieved by intramolecular interaction of the SH2 domain with the phosphorylated tyrosine 527 located in the C-terminal region. 43 In contrast, there is no internal phosphotyrosine–SH2 domain interaction in Abl, precluding this inhibitory mechanism. Instead, important inhibitory constraints are imposed on c-Abl both by the myristoylated cap and SH3 linker interactions (the latter are shared with Src). Adding complexity to this theme, through alternative splicing of exon 1, 2 N-terminal Abl variants can arise (myristoylated Abl 1b v. nonmyristoylated Abl 1a that lacks the sequence required for myristoylation through alternative splicing) (Fig. 1A and 1B). Differences in kinase regulation arise as the myristate moiety binds directly to the C-terminal lobe of the kinase domain and by the cap region phosphoserine 69, which binds to the SH2 domain. 44 These interactions further serve to lock the SH3-SH2 “clamp” onto the catalytic domain, predicting that Abl 1a is partially active or more activatable by SH2 ligands, which might lead to the “open” conformation44,45 more easily in Abl 1a than Abl 1b (Fig. 1C). Importantly, based on structural studies, in the elongated structure, integrity of the Abl SH2 domain–catalytic domain interaction is critical for maintaining Abl kinase activity. 45 In the “closed” Abl 1b form, SH2 domain binding to phosphopeptides prevents Abl kinase inhibition by the myristoyl group in cis. 41 The myristoyl group binding in trans,41,42 or small compounds mimicking its action,46,47 stabilizes the position of the C-terminal helix of the catalytic domain, αI, resulting in the inhibited conformation of the kinase. Therapeutically, this observation may have clinical value. An improved CML therapeutic compound called GNF-5 (or its subsequent generations) acts synergistically with imatinib or nilotinib to inhibit the Abl mutation, T315I, thus offering treatment of this imatinib-resistant mutation of BCR-Abl. 48 Taking into consideration the increasing role of Abl dysregulation in solid cancers, it will be important to understand the clinical significance of Abl kinase isoforms and their sensitivities to anti-Abl compounds.
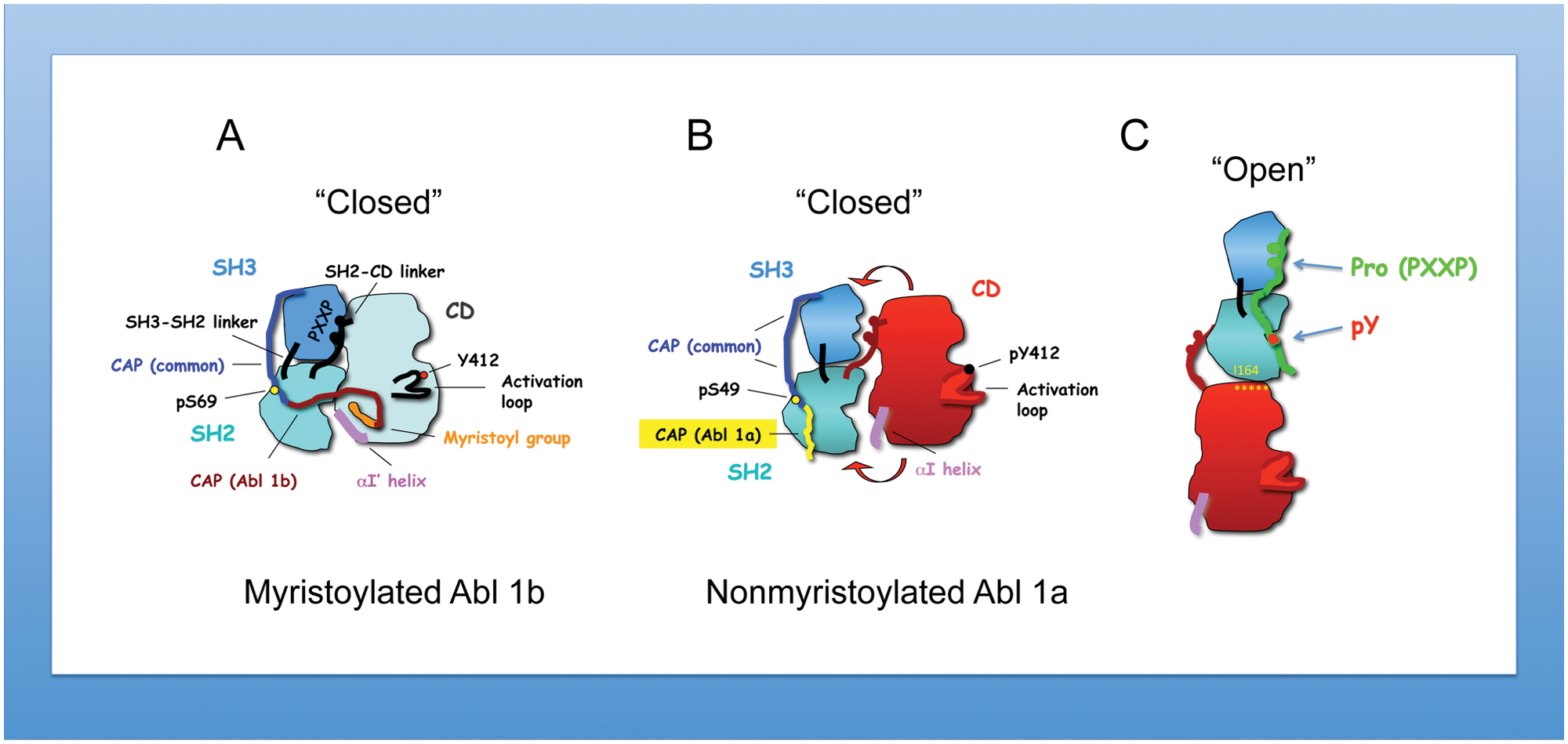
Structural differences of myristoylated versus nonmyristoylated Abl kinase. (
Modulation of Abl Activity by Abi1 and Crk
As described above, the SH2 and SH3 domains of Abl either maintain the autoinhibitory unit or facilitate their interaction with a wide range of adaptors and signaling proteins. When engaged to their respective ligands, this essentially unlocks the autoinhibited conformations, altering kinase activity and downstream tyrosine phosphorylation. While the nature of these interactions is complex and has been described in other excellent reviews in this monograph, here, we focus on the Crk and Abi1 proteins as an example of complexity in the regulation of Abl kinases and illustrate how stoichiometry may be controlled by substrate availability. As noted below, both Abi1 and Crk possess SH3 domains that bind to the Abl proline-rich domain (PRD) and appear to compete with each other to bind Abl.
Abi1
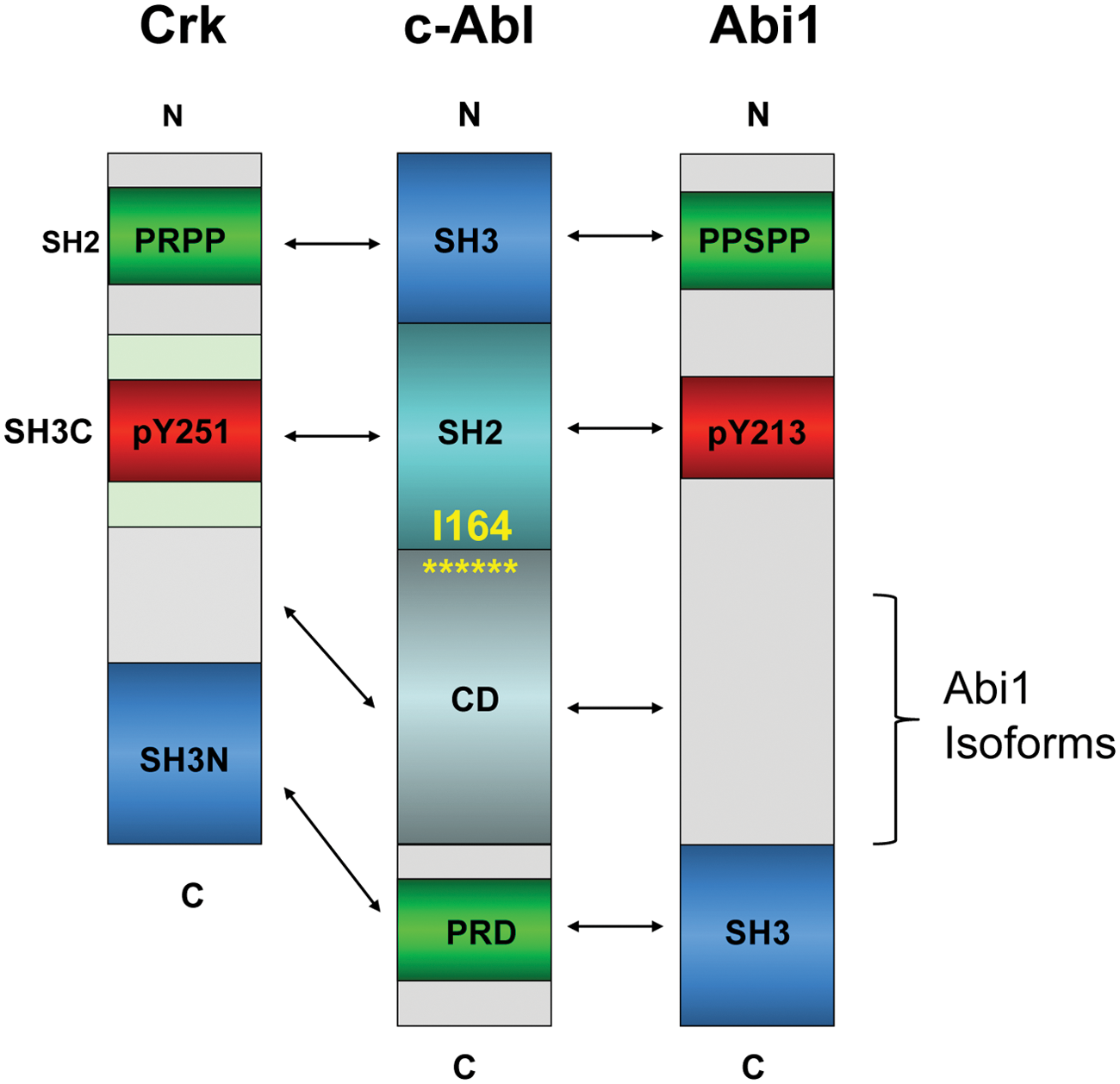
Abi1, together with Abi2 and Abi3 (also known as NESH), are a family of adaptor/scaffolding proteins that are originally characterized as Abl interactor proteins.49-52 The structure of Abi proteins is also generally conserved, characterized by 1 well-distinguished domain, the SH3 domain located in the C-terminus that binds type II proline peptides, including the PRD of Abl (Fig. 2). Juxtaposed to the SH3 domain as well as throughout the middle of Abi1, several proline-rich sequences including PEST and PXXP sequences are intercepted by tyrosine residues that are subject to posttranslational modifications. Relevant to the discussions here, several of these tyrosine residues are phosphorylated by Abl, which can potentially bind proteins with SH2 domains, including the Abl SH2 itself (Fig. 2) 53 and SH2 domains of other kinases such as p85–PI3 kinase and Src, which indirectly transmit signals downstream from Abl. 54
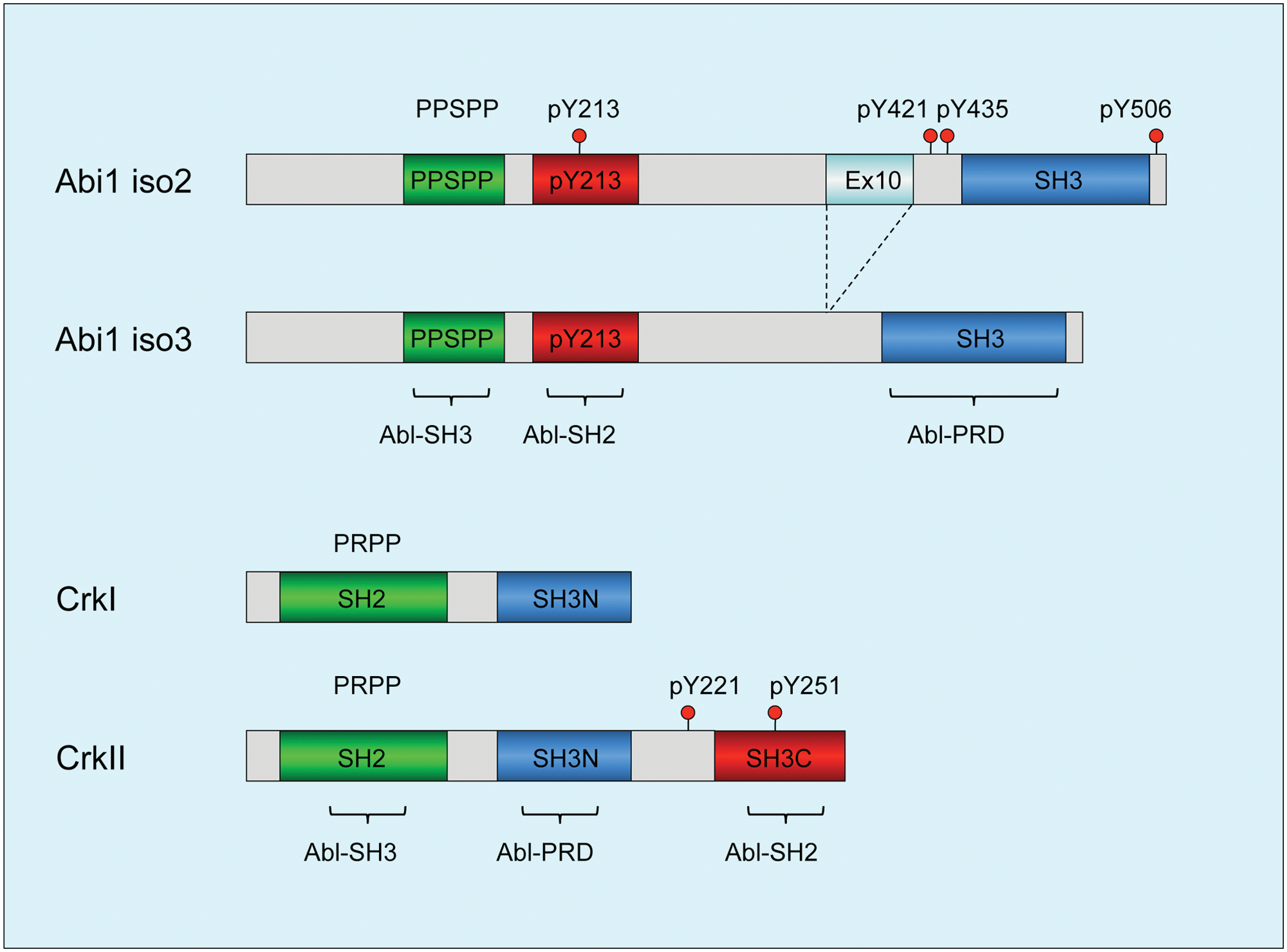
Diagrams depicting structural determinants of Abi1 and Crk: functional domains of Abi1 and Crk that regulate c-Abl tyrosine kinase. (
Intriguingly, the proximity of pY213 (Abl SH2) and PPSPP (Abl SH3) sequences on Abi1, along with SH3 domain binding to the PRD of Abl (this possibility was demonstrated for the conserved Abi2–SH3 domain), 55 predicts that Abi1 can interact with Abl using 3 independent motifs and when tyrosine phosphorylated would comprise a consolidated ligand-Abl SH3-SH2 domain binding surface possibly to act as a co-regulator of Abl function at multiple levels that operate cooperatively (Fig. 2). Cooperativity of the Abl SH3-SH2 interaction with ligands was previously suggested by Cowburn et al. 56
CT10 Regulator of Kinase: Crk
Crk, together with CrkL, is a family of adaptor proteins that lack tyrosine kinase activity but transmit intracellular signals downstream of tyrosine kinases. In vertebrates, Crk is alternatively spliced to produce 2 variants, crk I, composed of a SH2 and a SH3 domain, and a more abundant variant called crk II, which has an additional C-terminal SH3 domain and a 50–amino acid linker between the SH3 domains. 57 Crk-like (crk-L) is encoded by a distinct gene as crk and is structurally most similar to Crk II, although despite approximately 80% homology in the protein sequence in the SH domains, they have distinct nonoverlapping roles in development, reflected by the fact that knockouts are either embryonic (CrkL) or perinatal (Crk) lethal. Recent studies by Kalodimos and Inagaki 58 revealed fascinating distinctions in the SH2 and SH3 domain organizations between Crk II and CrkL, the latter acting as a preferred substrate to the Bcr-Abl oncoprotein. 59 Unexpectedly, these differences can be attributed to the distinct folding properties of Crk II and CrkL, mediated in large part by the interdomain regulatory elements.
At the functional level, the tandem SH2 and SH3 domains in Crk and CrkL recruit tyrosine-phosphorylated proteins and proline-rich proteins, respectively, and relevant to the arguments here, like Abi1, the first Crk SH3 domain (SH3N) binds to the PRD of Abl. While Abl has multiple PXXP motifs in its PRD, Antoku and Mayer 60 elegantly showed that they each contain sequence-specific information to bind distinct subsets of SH3 domains, although a comparison of the sequences for Abi1 and Crk was not directly investigated. Interestingly, in the study by Jung et al., 55 these investigators found that phosphorylation of Abl by PAK2 stimulated the tyrosine kinase activity of Abl in part by blocking the interaction with Abi1 and enhancing the interaction with Crk. In agreement with this model, as shown in Figure 3, Crk acts as a bona fide competitive inhibitor for Abi1 binding to Abl. This clearly supports the idea that Crk and Abi1 are in dynamic equilibrium for Abl, which will likely depend on 1) the concentration of Crk versus Abi1 in the cell, 2) their posttranslational modifications, and 3) the subcellular localization of Abl.
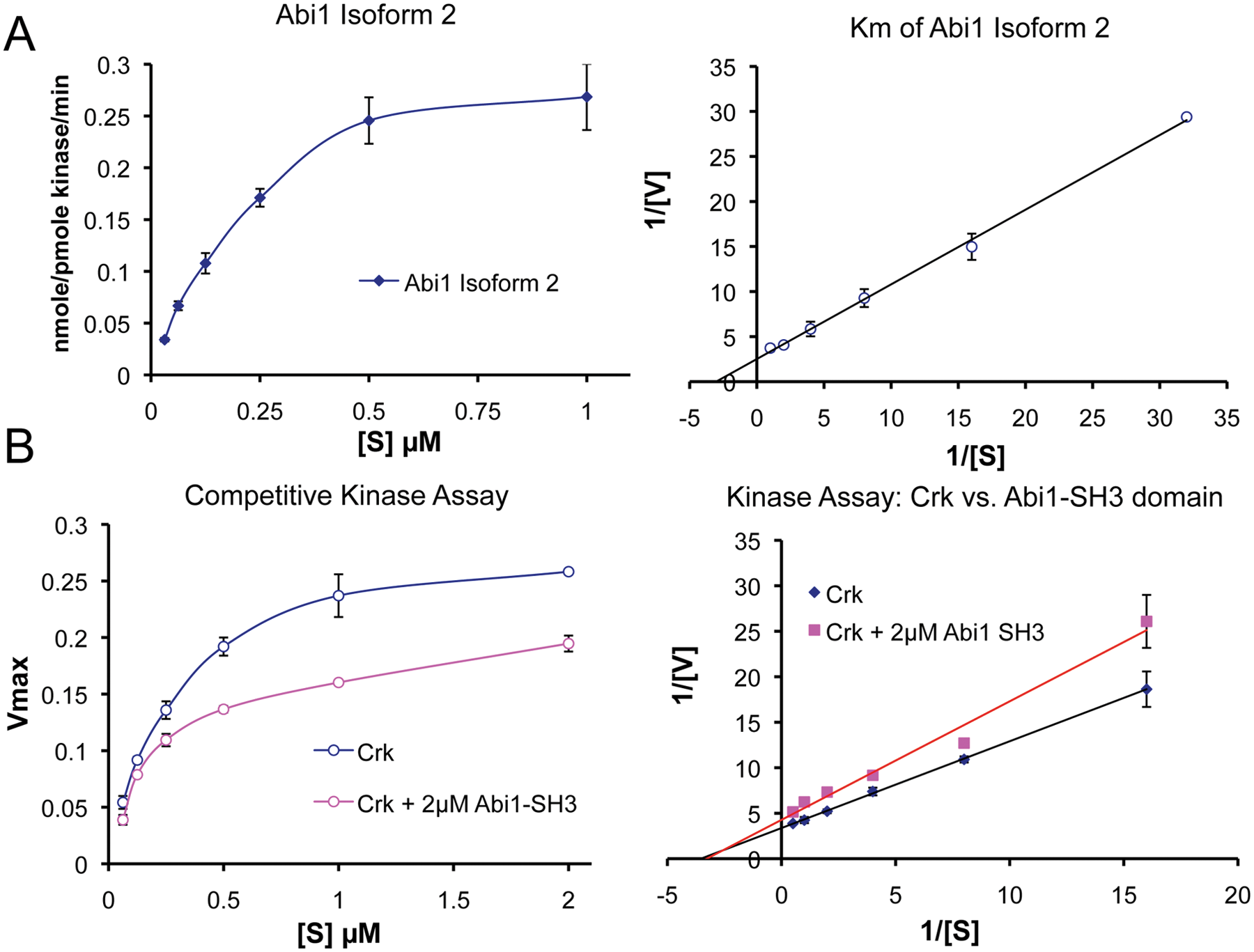
Abi1 competes with Crk at the SH3 domain interaction site. (
In addition to an apparent direct competition of Abi1 and Crk to 1 or more of the PXXP motifs in the PRD of Abl, Crk also appears to comprise a similar consolidated ligand for the Abl SH3/SH2 surface as Abi1. In this capacity, Sriram et al. 61 showed that EGFR induces phosphorylation of Crk on pY251, which in turn induces binding to the Abl SH2 domain. In combination with this observation, Anafi et al. 62 showed that Crk also binds to the Abl SH3 domain via PRPPVP within an extended loop in the SH2 domain. As shown in the model in Figure 4, these data suggest that Abi1 and Crk interact with conserved motifs, acting synergistically and possible cooperatively. These interactions include both Abi1 and Crk binding to the PRD of Abl, pY213 (Abi1) and pY251 (Crk) binding to the Abl SH2 domain, and PPSPP (Abi1) and PRPPVP (Crk) binding to the Abl SH3 domain.
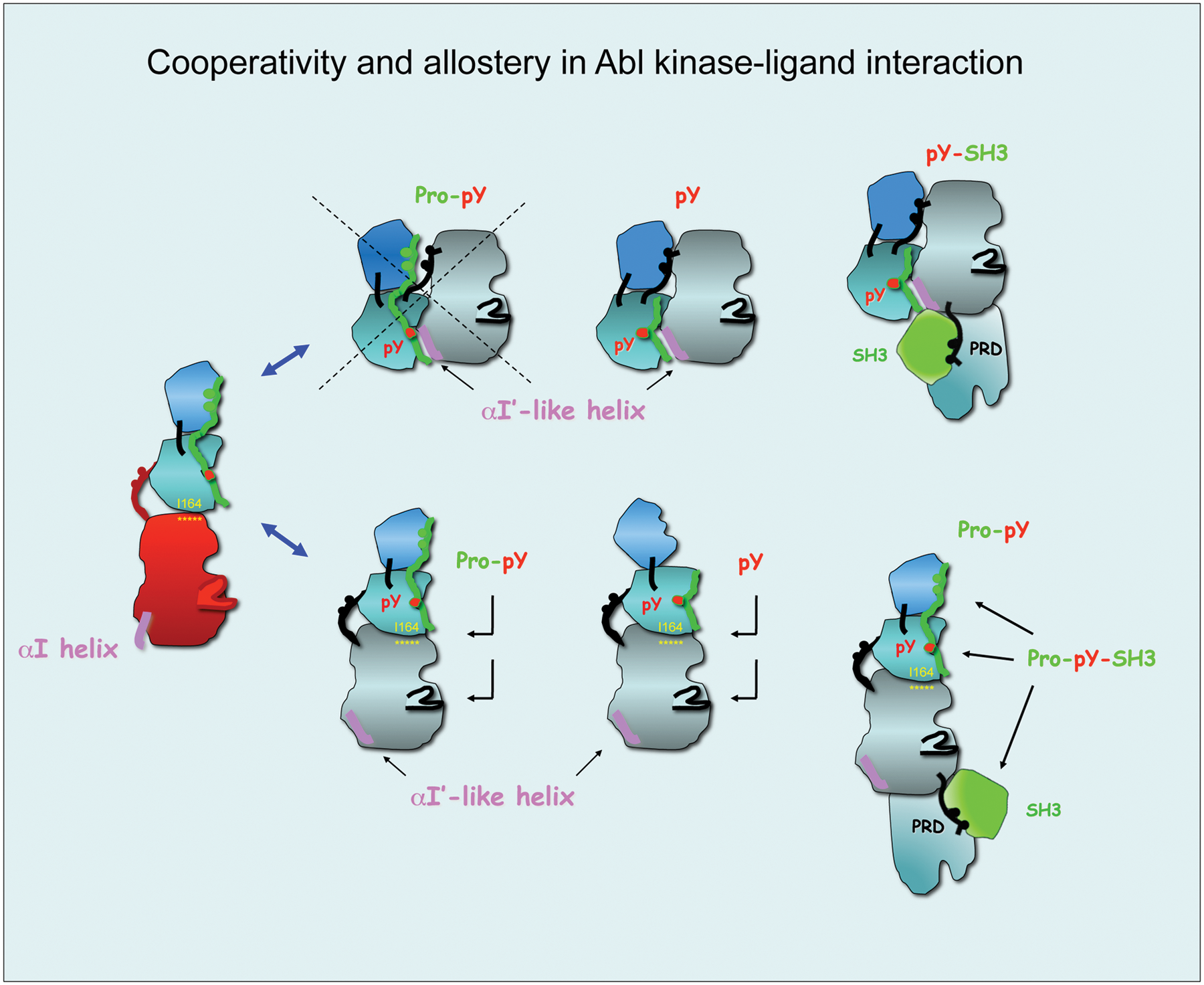
Cooperativity and allostery in Abl kinase-ligand interactions: candidate binding modes of Abi1 and Crk. Protein modules of Abi1 and Crk have the potential to interact with different Abl kinase conformations and to engage all 3 regulatory domains of Abl. (
Modes of Abl kinase activity regulation by Crk and Abi1 and their functional consequences: can adaptor proteins play a dual role in Abl activation and inhibition?
In the above arguments, we posit that Crk and Abi1 can compete for binding PRD motifs in Abl. Furthermore, once bound, important distinctions arise with respect to pY213 (Abl) and pY251 (Crk) binding to the SH2 domain of Abl. This is because the SH2 domain interaction with the Abl catalytic domain is proposed to play a dual role in Abl kinase activity: The SH2 domain inhibits Abl activity in the “closed” autoinhibited structure but promotes Abl activation in the “open” active conformation. By similar argument, ligand-mediated phosphotyrosine–Abl SH2 interaction has the potential to activate Abl activity by unlocking the closed conformation by disrupting the SH2–catalytic domain interaction, as proposed by Hantschel et al. 41 or by disrupting the active open “elongated structure,” as demonstrated by the loss-of-function mutation I164E in Abl 45 or by the anti-Abl SH2 domain antibody.63,64
Therefore, despite that both Abi1 and Crk each interact with Abl and appear to use overlapping regulatory mechanisms to engage the SH2 and SH3 domains of Abl, the biological consequences of Abi1 and Crk may be different, the former inducing Abl inhibition and the latter inducing Abl transactivation. For example, we have shown that Abl 1a can be inhibited when Abi1 binds in trans to the proline-rich region of Abl. As a consequence of this interaction, Abi1 becomes phosphorylated on Tyr213, which in turn induces an interaction with the Abl SH2 domain. What is perplexing from this finding is why Abl SH2 domain engagement by pTyr213 Abi1 does not result in Abl transactivation. As noted above, an analogous interaction between Abl and Crk causes Tyr251 phosphorylation; the latter also binds the Abl SH2 domain but, unlike Abi1, causes Abl activation. One possible scenario, which at present remains speculative, is that the Abl-pTyr213 Abi1 complex adopts an autoinhibitory complex in trans, similar to the Src-pTyr527 intramolecular interaction. This is consistent with our observations that Y213F Abi1 is much less effective in the transinhibition of Abl by Abi1. Under this model, the Abi1 PPSPP would not be able to engage the SH3 domain interacting with the Abl catalytic domain in the autoinhibited/closed conformation (Fig. 4).
However, another intriguing possibility for how and why Abi1 inhibits Abl may also result if indeed Abl is in further equilibrium with an open Abl conformation and includes an effect on the integrity of the SH2–catalytic domain interaction (Fig. 5). Here, there might be a role for the earlier mentioned alternatively spliced region of Abi1. Clearly, the use of solution phase nuclear magnetic resonance spectroscopy could be used to resolve these questions and provide more formal proof for the proposed snap-lock structure alluded to above or for the effect on the I164 residue interaction with the catalytic domain. Destabilizing mutations in the SH2–catalytic domain interaction were found in several kinases to associate with various human cancers including lung cancer (Fer kinase), X-linked agammaglobulinemia (Btk), and severe combined immunodeficiency (Zap70 and Jak3). 65
Abi1 and Crk might regulate Abl kinase activity by affecting the stability of the SH2–catalytic domain (CD) interaction in the open conformation of Abl kinase. In our article, 53 we propose that Abi1 Pro-pY213 peptide inhibits Abl kinase activity through binding to Abl SH3-SH2 in an elongated conformation as proposed by small-angle X-ray scattering measurements 44 and affecting the integrity of the SH2-CD interaction. pY213 peptide might exert its inhibition by binding to the Abl SH2 domain only. Based on our cell studies 53 indicating that Abi1 inhibits c-Abl kinase, we propose that the mechanism involves multiple interactions of Abi1 and c-Abl at multiple domains starting from Abi1 PPSPP and pY213 sequences through the middle region of the Abi1 protein and including the SH3 domain at the C-terminus. The Abi1 SH3 domain was previously demonstrated to interact with the Abl proline-rich domain (PRD) 49 ; homologous Abi2 SH3 was also demonstrated to interact with the Abl PRD.51,55 In fact, the “open” elongated conformation of Abl is compatible with the positioning of Abl and Abi1 binding sites alongside each other with the SH3 domain of Abi1 binding to the proline-rich region of Abl located C-terminally to the CD. Crk is proposed to interact in a similar fashion. The role of Abi1 or Crk on Abl kinase activity will depend on the effect on SH2-CD interaction. The critical I164 is indicated in yellow. Brackets indicate the alternatively spliced region of Abi1.
Interestingly, in the extended open conformation, the Abl catalytic domain is located in between the SH3 domain and PXXP-pY binding sites; thus, it is reasonable to hypothesize that Abi1 or Crk binding might also involve interactions with the Abl catalytic domain. It will be interesting to learn whether these sequences have any effect on Abl kinase activity. In the case of Abi1, the sequences located between pY213 and the SH3 domain represent the differentially spliced region of Abi1. 52
What is the biological significance of the binary regulation of Abl by Abi1 and Crk in human cancers, and what are the consequences for actin cytoskeleton regulation?
Dysregulation of Crk in cancers is well established 66 Lung,67,68 breast,69,70 and gastric cancers 71 as well as glioblastoma 72 have demonstrated mostly elevated levels of Crk 66 (see Bell and Park in this monograph). At the mechanistic level, both transcriptional and posttranscriptional mechanisms may contribute to Crk expression. In the latter scenario, a Crk-specific miRNA (miR-126) is downregulated in several cancers, which leads to elevated Crk levels 71 (see Tsuda and Tanaka in this issue).
However, in the case of Abi1, the dual role of Abi1 function is observed in human cancer (Table 1). Increased Abi1 is linked to enhanced oncogenesis, for example, in invasive breast cancer, ovarian cancer, and leukemia and to tumor suppression in gastric 73 and prostate cancers.74,75 Low levels of Abi1 are consistent with the tumor suppressor hypothesis in prostate and gastric cancers. The proposed binary regulation of c-Abl tyrosine kinase by Crk competing with Abi1 for c-Abl might help to explain the dysregulation of actin cytoskeleton dynamics.
Role of Abi1 in Cancer
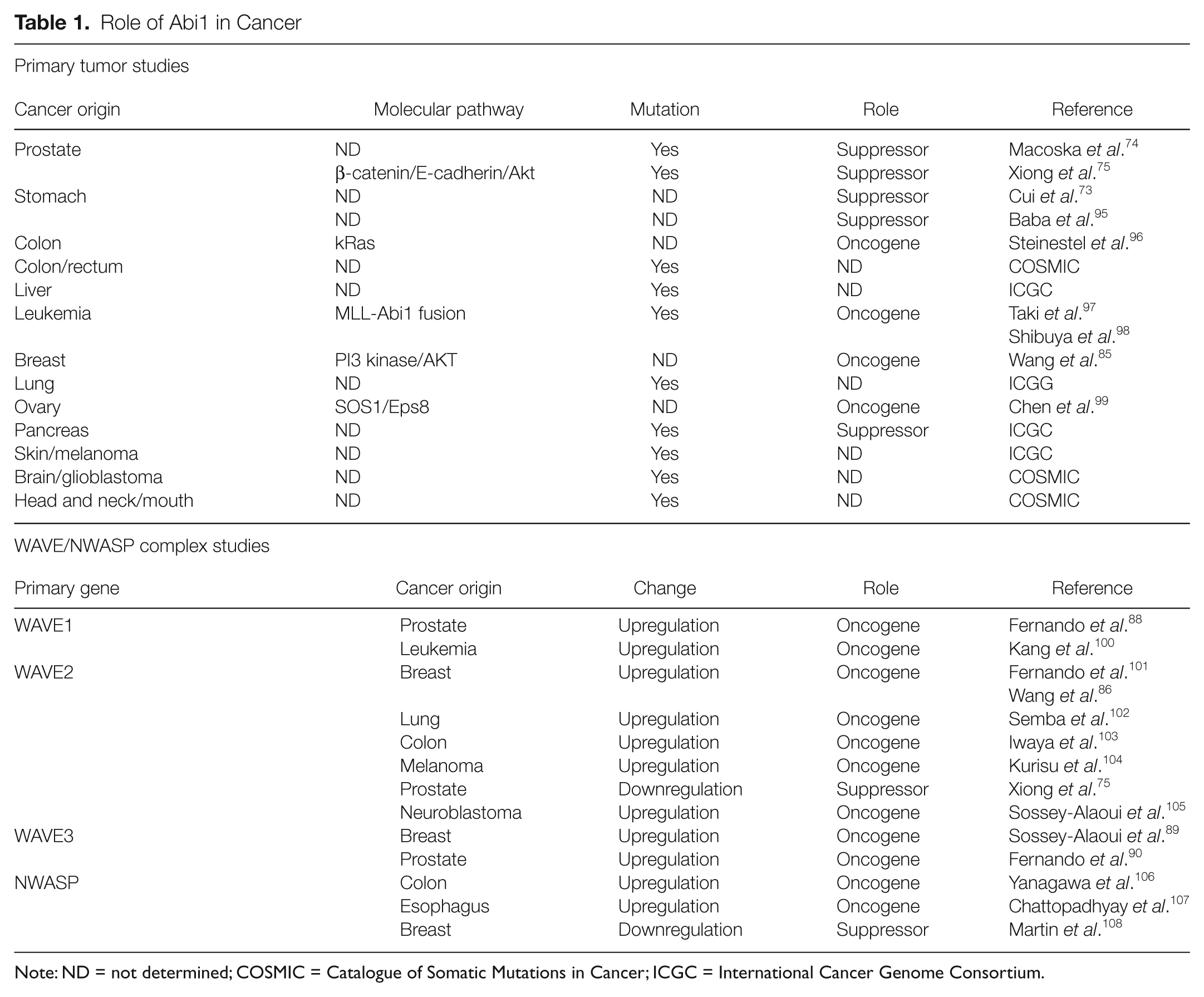
Note: ND = not determined; COSMIC = Catalogue of Somatic Mutations in Cancer; ICGC = International Cancer Genome Consortium.
But what could be the consequences of altered Crk/Abi1 ratios in cancer? Abi1 is an established component of actin regulatory complexes WAVE and NWASP; it also enters the p85 regulatory subunit of PI3 kinase54,76 and can target the Src family of tyrosine kinases through pY421–SH2 domain interaction. 54 High levels of Crk opposite Abi1 would likely compete off Abi1 from Abl, thus leading to higher levels of Abi1 in its actin regulatory complexes and leading to dysregulation of the actin cytoskeleton and resulting in tumorigenic phenotypes in cancer. These consequences of Abi1 level dysregulation are listed in Figure 6.
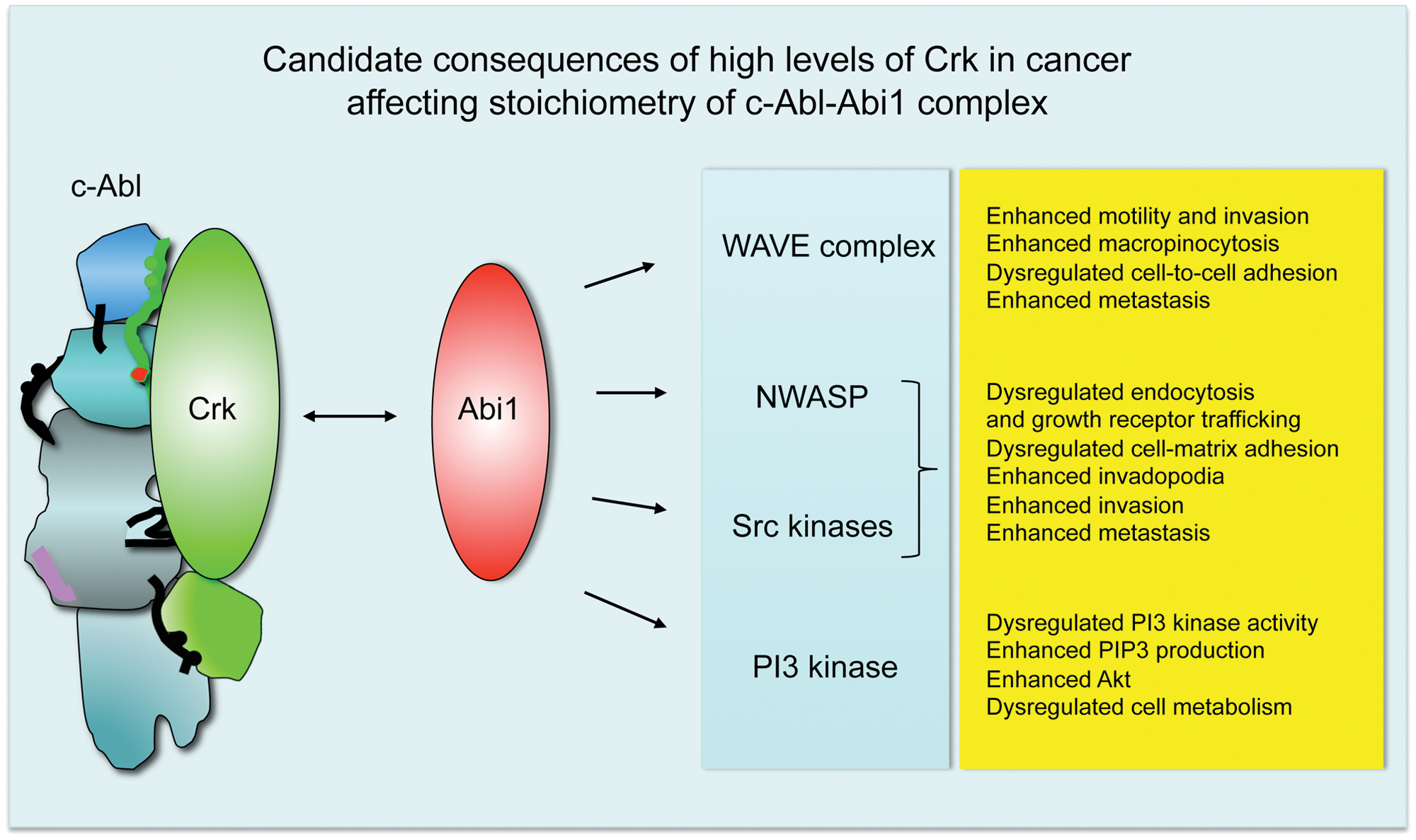
Dysregulated stoichiometry of Crk-Abl and Abi1-Abl complexes leads to an invasive phenotype of the tumor-associated actin cytoskeleton. Biochemical data49,53,61,62 indicate that Abi1 and Crk target the same regulatory domains of c-Abl, thus suggesting binary regulation. We propose that Crk binding to Abl would lead to increased levels of Abi1 in actin regulatory complexes such as WAVE and/or NWASP, which would likely result in a tumorigenic phenotype (
Furthermore, enhanced WAVE complex activity leads to enhanced cellular motility and invasiveness. Dysregulation of WAVE complexes might lead to abnormal cellular adhesion and dysregulation of adherens junction formation, as suggested by the loss of CYFIP in several epithelial cancers 77 and our mouse Abi1 knockout studies. 75 Enhanced levels of Abi1 WAVE can also explain the enhanced cell–extracellular matrix adhesion, leading to the progression of leukemia78,79 and breast cancer. 80 WAVE complex dysregulation in cancer has been elegantly covered in a recent review by Kurisu and Takenawa. 81 Enhanced invadopodia formation is a feature of breast cancer in vivo and in vitro through activation of Src80,82 and NWASP.83,84
Higher levels of Abi1 might lead to PI3 kinase upregulation through SH2 domain interaction as suggested54,76 and subsequent metabolic changes as a consequence of enhanced PIP3 and Akt signaling as described, for example, for invasive breast cancer.85,86 In prostate cancer, downregulation of the PI3 kinase α isoform but upregulation of the PI3 kinase β isoform are associated with invasive changes. 87 The role of Abi1 in PI3 kinase isoform regulation is yet to be determined, but it might be important from the clinical point of view of the widespread evaluation of PI3 kinase inhibitors in solid cancer trials and the critical role of Abi1/WAVE/NWASP complexes in tumor invasion.
High levels of Crk versus high levels of Abi1 in some cancers can also cause higher on and off rates of the proteins interacting with Abl, PI3 kinase, and Abi1-containing actin regulatory complexes. For example, enhanced Abi1 and Crk could lead to an enhanced turnover of invadopodia, breaking up the matrix and leading to very invasive changes.
While the preceding discussion mainly focused on the stoichiometry of Abi1 and Crk, more recently, it has also been shown that mutations can occur in Abi1, adding complexity to the aforementioned themes relating to stoichiometry. Mutations in Abi174,75 (Table 1) can also affect the dynamics of the binary Abi1-Crk regulation of Abl. The prostate tumor LNCaP cell line contains the loss-of-function mutation in the Abl SH2 domain binding site of Abi1, which lacks pY213, 74 leading to dysregulation of Abl kinase activity and enhanced phosphorylation of Crk. 53 Loss of Abi1 in prostate cancer mouse models leads to decreased cell-to-cell adhesion through downregulation of β-catenin and E- cadherin downstream from the WAVE2 complex. 75 In humans, upregulation of the other WAVE complexes such as associated with WAVE1 88 or WAVE389,90 underlies invasive prostate tumors. Hence, depending on the ability of mutated Abi1 to reconstitute WAVE complexes, and to compete with Crk for Abl as proposed here, invasiveness might be associated with WAVE complex dysregulation, as observed in cells lacking Abi1. 91 Dysregulation of WAVE complexes is not only specific to prostate cancer: elevated levels of WAVE3 are found in invasive breast cancer. This is also true for WAVE2, which is also upregulated in invasive lung and colon cancer as well as in melanoma (Table 1).
The devil is in the detail: complexity versus simplification
Over the past several decades, much effort has been delineated in the role of tyrosine kinases in cancer. Now, it is equally apparent that analogous to kinases, modulation of adaptor proteins can have equally important roles in cancer and may do so in part by modulating tyrosine kinase signaling. As in many forums in biology, information learned from one area is often inexplicably linked to other systems. As we turn to explain the role of Abl regulators such as Abi1 and Crk in a simple analysis, one might require more detailed information from this model system. This might lead to better explanations of tumor progression mechanisms, drug action mechanisms, and their differential efficacies in different types of cancers.
Footnotes
Acknowledgements
The authors thank Stephan Knapp (Oxford University, Oxford, UK), David Cowburn (Albert Einstein College of Medicine, Bronx, NY), and Tony Koleske (Yale University, New Haven, CT) for helpful discussions.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: L.K. was supported by grants from the Department of Defense Prostate Cancer Research Program (W81XWH-08-1-0320), the National Institutes of Health (R01 NS044968), and the FM Kirby Foundation Inc. R.B.B. was supported by grant R01-CA165077 from the National Cancer Institute.