
Abstract
Abl kinases are prototypic cytoplasmic tyrosine kinases and are involved in a variety of chromosomal aberrations in different cancers. This causes the expression of Abl fusion proteins, such as Bcr-Abl, that are constitutively activated and drivers of tumorigenesis. Over the past decades, biochemical and functional studies on the molecular mechanisms of Abl regulation have gone hand in hand with progression of our structural understanding of autoinhibited and active Abl conformations. In parallel, Abl oncoproteins have become prime molecular targets for cancer therapy, using adenosine triphosphate (ATP)–competitive kinase inhibitors, such as imatinib. Abl-targeting drugs serve as a paradigm for our understanding of kinase inhibitor action, specificity, and resistance development. In this review article, I will review the molecular mechanisms that are responsible for the regulation of Abl kinase activity and how oncogenic Abl fusions signal. Furthermore, past and ongoing efforts to target Abl oncoproteins using ATP-competitive and allosteric inhibitors, as well as future possibilities using combination therapy, will be discussed.
Structure and Regulation of Abl
The Abl family of cytoplasmic tyrosine kinases consists of 2 members, Abl and Arg (Abl-related gene), encoded by the ABL1 and ABL2 genes in humans, and has important roles in various biological processes.1,2 Abl kinases share a central SH2-kinase domain unit with the majority of other cytoplasmic kinases 3 and have a long C-terminal tail, termed the last exon region, that carries numerous protein-protein interaction sites (Fig. 1). 4 The activity of Abl kinases is regulated by a complex set of intramolecular interactions that impinge on the Abl kinase domain and lead to effective inhibition of tyrosine kinase activity both in vitro and in vivo. Even a partial, albeit persistent, disruption of autoinhibitory constraints results in oncogenic transformation. Kinase activity of the full-length Abl protein is low in vitro and hard to detect in unstimulated cells. In contrast, the isolated Abl kinase domain has a 10- to 100-fold higher kinase activity than the full-length protein (unpublished observation). This indicates that the additional domains present in Abl kinases mediate both intra- and intermolecular interactions that either directly or indirectly dampen catalytic activity of the kinase. The SH3 and SH2 domains of Abl play key roles in mediating autoinhibition and will therefore be discussed first.
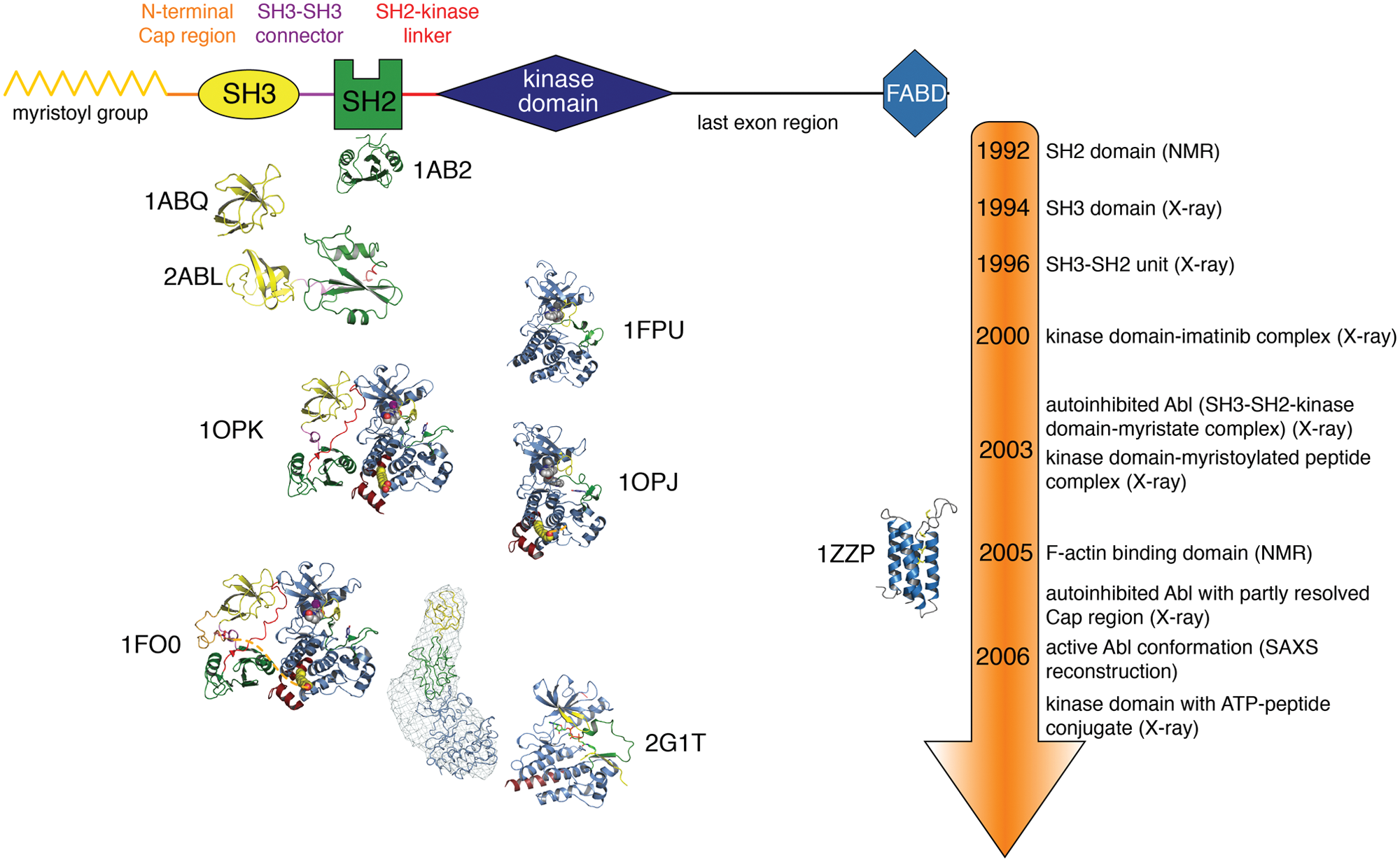
Timeline of Abl structural insights. A schematic domain representation of Abl is shown on top of the figure. Below, the structures, staggered by the time when they were published, of the different Abl domains and domain combinations are shown in cartoon representation. The structures are colored as in the schematic domain representation on top. Once different structures of the same domains (e.g., obtained with different methods, different crystal forms, mutants, drugs) were published simultaneously, only one representative structure is shown for graphical convenience. The PDB entries from which the representations were derived are shown next to the respective structure.
Abl SH3 and SH2 Domains
SH3 and SH2 domains are, with more than 300 and 120 members, respectively, among the most common modular protein-protein interaction domains found in human proteins. 5 SH3 domains bind to peptides forming polyproline type II helices, whereas SH2 domains bind to phosphotyrosine-containing peptides. The Abl SH3 domain was the first SH3 domain that was crystallized with a bound ligand peptide and therefore revealed how the SH3 domain mediates protein-protein interactions (Fig. 1). 6 Likewise, the Abl SH2 domain was the first SH2 domain to be structurally characterized and gave important insight into the folding and dynamics of the domain (Fig. 1). 7 Based on these hallmark structures, fundamental principles of ligand recognition, specificity, and relation to other protein-protein interaction domains were made in the following years. 5 The structural elucidation of the Abl SH3-SH2 dual domain construct indicated flexible linkage of the 2 domains without major interactions (Fig. 1). 8
Abl Autoinhibition by Its SH3 and SH2 Domains
It could be conclusively demonstrated that Abl is autoinhibited, and no other proteins that may serve cellular inhibitors need to be envisaged. 9 A major role in mediating autoinhibition is attributed to the Abl SH2 and SH3 domains. Elegant biochemical work demonstrated an intramolecular interaction of the Abl SH3 domain with its own SH2-kinase domain linker. This sandwiches this linker between the SH3 domain and the N-terminal lobe of the kinase domain (Fig. 2). Perturbation of this network of interactions strongly activated Abl kinase activity. 10 The SH2-kinase linker adopts the conformation of a polyproline type II helix, which is the preferred ligand of the SH3 domain. This mechanism is conserved in the Src kinases, which share the same domain organization and a high sequence identity in the folded domains and domain linkers with the Abl kinases.
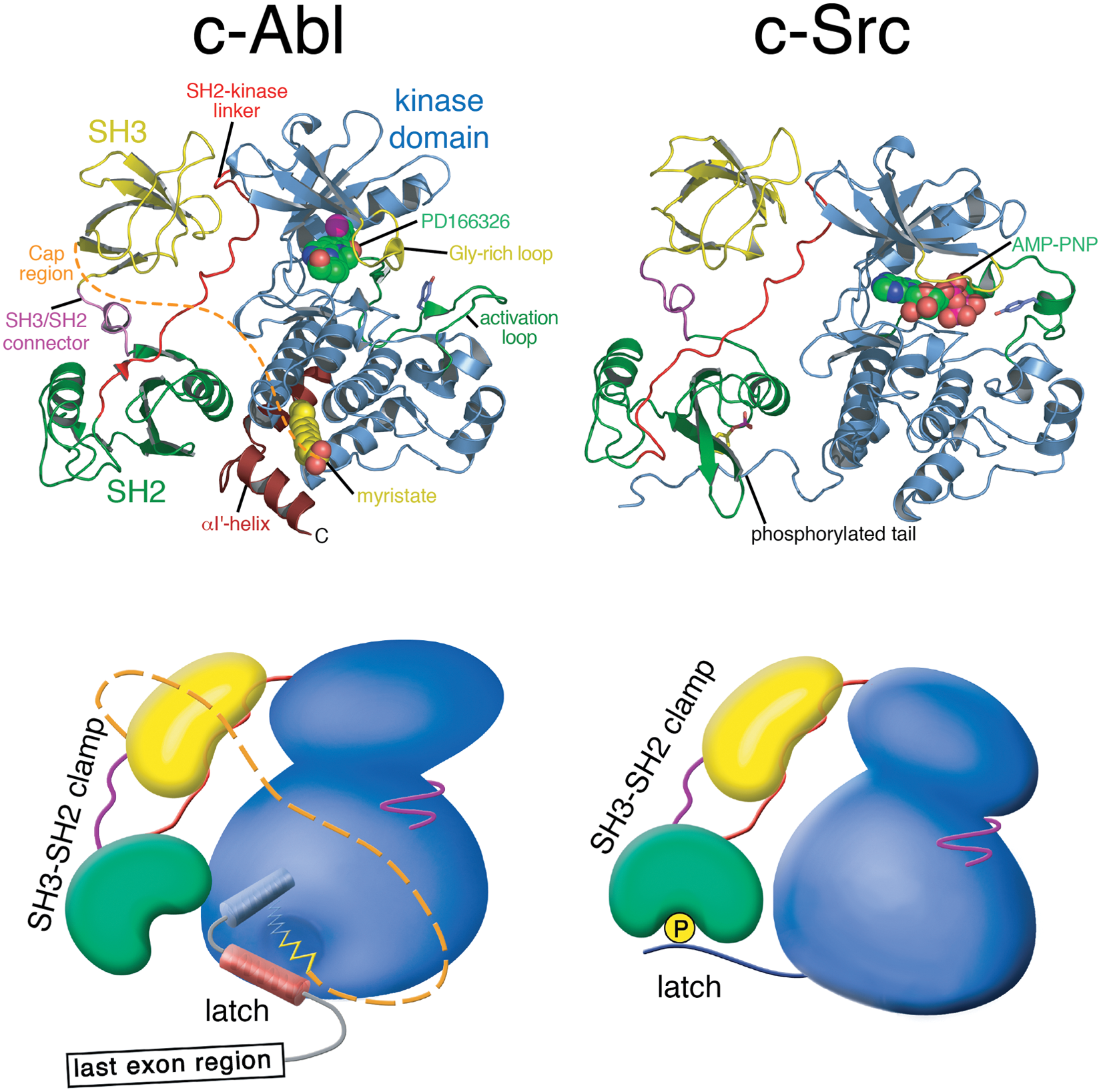
Structure of autoinhibited Abl and Src. Cartoon representation of autoinhibited Abl in complex with the adenosine triphosphate (ATP)–competitive inhibitor PD166326 (left; PDB entry 1OPK, 18) compared with autoinhibited Src in complex with the ATP analogue AMP-PNP (right; PDB entry 2SRC, 14). Below the cartoon representations of the crystal structures, more schematic representation are used that should illustrate global conformation differences of the 2 kinases. In both kinases, the SH3-SH2 domain unit forms a clamp that inhibits the kinase domain. In Src, the tyrosine-phosphorylated tail binding to the SH2 domain latches the clamp. The myristoyl group of Abl serves as a latch for the SH3-SH2 clamp by inducing a conformational switch in the C-terminal kinase domain helix that gates clamp binding.
In contrast to the SH3 domain, the role of the SH2 domain in regulating Abl activity remained unclear much longer and turned out to be very different from Src kinases. 11 The Src SH2 domain binds to the C-terminal tail of the kinase domain that is phosphorylated on a single tyrosine residue by the Csk kinase and thereby keeps Src in a closed/assembled conformation of low catalytic activity (Fig. 2).12-14 The long C-terminal last exon domain of Abl lacks a functional equivalent of the C-terminal phosphotyrosine in Src. Autoinhibted Abl is not phosphorylated on tyrosine residues, showing that the SH2 domain does not bind an intramolecular ligand. In addition, the analysis of SH2 domain deletions or mutations in Abl and the oncogenic fusions v-Abl and Bcr-Abl did not unequivocally decipher the function of the SH2 domain for kinase activity and transformation.15-17 The determination of crystal structures of autoinhibited Abl and accompanying functional experiments resolved most of this nebulosity.18,19 The Abl SH2 domain forms an extensive interaction interface with the C-terminal lobe of the kinase domain that is stabilized by an interlocking network of hydrogen bonds (Fig. 2). 18 Due to the different relative orientation of the 2 kinase lobes in Src and Abl, the SH2 domain approaches the kinase domain much closer in Abl (Fig. 2). The SH2–C-lobe interface in autoinhibited Abl partly occludes access of phosphotyrosine ligands to the phosphotyrosine binding pocket (Fig. 2). 18 This positioning also explains why phosphotyrosine ligands and high-affinity engineered Abl SH2 binding proteins were able to stimulate kinase activity in vitro and in cells.19,20 Most important, docking of the SH2 domain to the C-lobe of the kinase is gated by the N-terminal myristate moiety that is bound to its binding pocket in the C-lobe.
Abl Regulation by Myristoylation
Abl and Arg have 2 alternatively spliced first exons, termed 1a and 1b in humans (type I and IV in mouse). The 1b splice form is 19 amino acids longer than Abl 1a and carries a myristate group, a saturated fatty acid with 14 carbon atoms, at its N-terminus. Protein N-myristoylation is implicated in targeting proteins to membranes, but myristoylation alone is not sufficient for stable membrane binding. Dual myristoylation and palmitoylation or additional polybasic amino acid stretches that interact with negatively charged phospholipids at the inner leaflet of the plasma membrane are necessary for membrane targeting. 21 In Abl 1b, neither of those 2 additional membrane targeting signals are observed, and in line with this, only a minor fraction of Abl is localized at membrane-proximal sites. Overall, Abl has diverse localizations in the cytoplasm, nucleus, and a variety of intracellular organelles (reviewed in Pendergast 2 ). Furthermore, nonmyristoylated Abl was not differentially localized than the myristoylated protein. 19 On the other hand, mutants defective in F-actin binding depleted membrane co-localized Abl, indicating that binding to the membrane-proximal cortical F-actin cytoskeleton rather than myristoylation is a major determinant of membrane localization. 22
In contrast, Abl myristoylation was found to be involved in regulating kinase activity. Mutants of Abl 1b that lack the myristoyl group show strongly deregulated cellular and in vitro tyrosine kinase activity. 19 A crystal structure of the kinase domain in complex with a myristoylated peptide corresponding to the very N-terminus of Abl 1b showed that the myristoyl group is buried in a deep hydrophobic pocket in the C-lobe of the kinase (Figs. 1 and 2). 18 Myristoyl binding to this pocket causes a 90° bending of the C-terminal α-I helix of the kinase domain. Only this event creates the complete docking site for the SH2 domain on the C-lobe and enables the assembly of the autoinhibited conformation of Abl 1b (Fig. 2). Mutations that block access to the myristate pocket strongly increase kinase activity. 19 Importantly, compounds binding to the myristate pocket act as allosteric Abl inhibitors (see below and reviewed in Hantschel 23 ).
Kinase Domain Structures
Studies on the structure of the Abl kinase domain revealed important insight into the regulation of catalysis and recognition mode of Abl kinase inhibitors. Early work showed that Tyr-412 in the activation loop is a major autophosphorylation site and constitutes a switch between the inactive and active kinase conformation.24,25 Co-crystal structures of the kinase domain in complex with imatinib and other kinase inhibitors exemplified binding modes of drugs and associated conformational changes in the kinase domain (reviewed in O. Hantschel, F. Grebien, and G. Superti-Furga, unpublished data; Fig. 1).26,27 Importantly, these structures were indispensable tools to rationalize the mechanism of action of point mutations causing drug resistance. 28 Structures in complex with adenosine triphosphate (ATP)–peptide conjugates showed a close structural resemblance to the inactive Src kinase domain (Fig. 1). 29 This conformation, termed Src-like inactive conformation, together with additional crystal structures and molecular dynamics simulations exemplified conformational dynamics of the wild-type and mutant Abl kinase domain and its consequences for drug binding and specificity over the closely related Src kinases.29,30
Active Abl: The SH2-Kinase Interface
Upon activation, Abl undergoes extensive domain rearrangements. One hallmark change is that the SH2 domain does not bind to the C-terminal lobe any more but forms an extensive interface with the N-terminal lobe of the kinase domain.31,32 These intramolecular interaction interfaces in autoinhibited Abl and active Abl involve different surfaces of the SH2 domain. The positioning of the SH2 domain on the N-lobe mediates allosteric activation of the kinase domain that is independent of its phosphotyrosine binding capability. This mechanism was also demonstrated in great structural and biochemical details for the tyrosine kinase Fes. 32 Furthermore, indirect evidence indicated that the SH2 domain in other cytoplasmic tyrosine kinases might also act as an allosteric activator, in line with the overall conservation of the SH2-kinase domain unit in these tyrosine kinases. Most important, the SH2-kinase domain interface in the oncogenic fusion Bcr-Abl was recently shown to be essential for leukemogenicity and represent a novel allosteric target for pharmacological intervention. 33
In addition to its allosteric regulatory role, the positioning of the SH2 domain on the N-lobe facilitates multisite (processive) phosphorylation of Abl substrates with multiple phosphorylation sites by binding to prephosphorylated (primed) substrates. 34 Mutation of the phosphotyrosine-binding pocket or its blockade by a high-affinity engineered protein antagonist impairs processive phosphorylation of the Abl substrate paxillin to the same extent as mutation of the SH2-kinase domain interface.20,33 It is important to note that the substrate specificity of the Abl kinase domain is very similar to the ligand-binding preference of the Abl SH2 domain, which not only indicates the co-evolution of the 2 domains but also rationalizes the above-described mechanism.34,35
Mechanisms of Abl Activation
In addition to their role as intramolecular regulators of kinase activity, intermolecular binding of the SH3 and SH2 domains to their respective ligands in a variety of interacting proteins and substrates is disrupting the inhibitory interactions. This appears to be a widely used mechanism of Abl activation. 4 Likewise, phosphorylation of Abl by upstream kinases or autophosphorylation events lead to conformational changes that disrupt the intramolecular engagement of the SH3 and SH2 domains and trigger the formation of intermolecular protein-protein interactions. 4 A well-documented example for this type of mechanism is phosphorylation of Tyr-245. 25 As described above, the SH2-kinase domain linker in Abl contains a PxxP motif and is bound by the SH3 domain. The second proline residue of the PxxP motif is replaced in Abl by Tyr-245, and its phosphorylation was predicted to disrupt the autoinhibited structure, consistent with the high levels of activity observed upon phosphorylation of Tyr-245 in Abl. 18
Abl Fusion Proteins in Cancer
c-Abl was discovered as the cellular homologue of the viral oncoprotein v-Abl that is expressed by the Abelson murine leukemia virus. 36 In humans, Abl kinases are involved in a number of chromosomal abnormalities in different cancers that lead to the expression of fusion proteins, but no (activating) point mutations in the ABL1 or ABL2 genes have yet been identified in human cancers or other diseases. In all human Abl fusion proteins, as well as in murine v-Abl, regions upstream of the Abl kinase domain are replaced by another protein (Fig. 3). The fusion partner contributes sequences that drive dimerization/multimerization of the kinase, which, by itself, was shown to trigger Abl activation. 37 In general, the fusion event leads to a loss of kinase autoinhibition by removing the myristoylation site and, in some cases, the SH3 and SH2 domains (Fig. 3).10,19 In addition to the gain in tyrosine kinase activity, Abl fusion partners trigger the activation of the oncogenic pathways. I would like to provide a summary of the structure and signaling of the most common Abl fusion proteins that are expressed in hematological malignancies. I will first focus on Bcr-Abl, as this is by far the most intensively studied and best understood among the Abl oncoproteins.
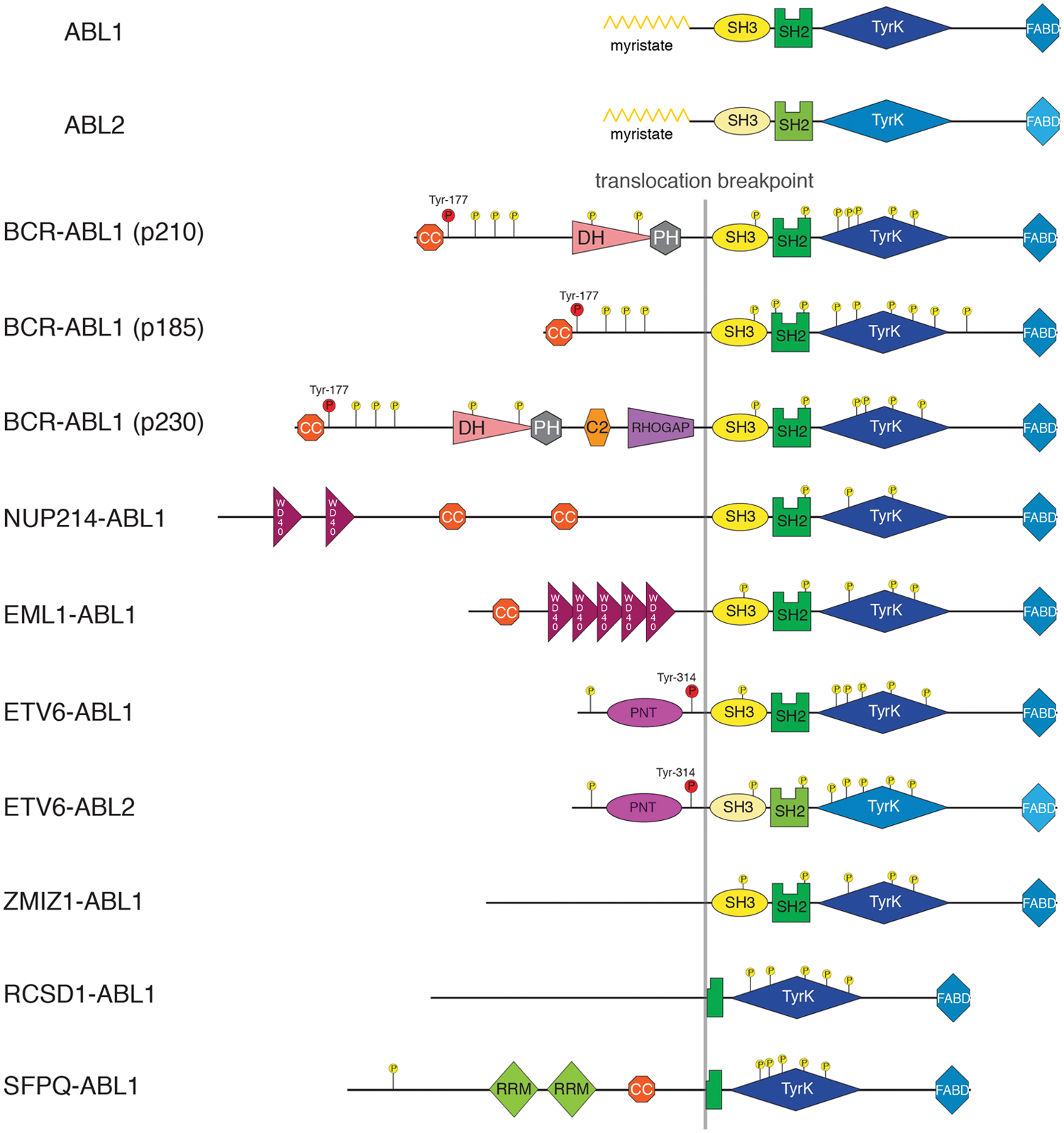
ABL1 and ABL2 fusions in cancer. Schematic domain representation of the proto-oncogene products of the ABL1 and ABL2 genes, as well as the known fusion gene products that mainly arise through chromosomal translocations. Known phosphorylation sites are indicated. The phosphorylation events on Tyr-177 in Bcr-Abl isoforms and Tyr-314 in Etv6-Abl that are discussed in more detail are indicated in red. Sizes of the proteins are approximately to scale. Domain abbreviations are as follows: TyrK, tyrosine kinase; FABD, F-actin binding; CC, coiled-coil; DH, Dbl-homolgy; PH, Pleckstrin-homology; RHOGAP, Rho GTPase-activating protein; WD40, Trp-Asp (W-D) terminating domain of ~40 amino acid length; PNT, pointed dimerization domain; RRM, RNA recognition motif.
Bcr-Abl
The t(9;22)(q34;q11) chromosomal translocation that results in the formation of the Philadelphia (Ph) chromosome was the first consistent chromosomal aberration linked to human cancer 38 (reviewed in Rowley 39 ). Seminal work in the following 2 decades showed that the Ph chromosome encoded the Bcr-Abl protein that is formed by the fusion of the ABL1 gene (on chromosome 9) and the breakpoint cluster region gene (on chromosome 22). The translocation breakpoint on chromosome 9 upstream of exon 2 of the ABL1 gene leads to loss of expression of the first exon of ABL1 in the Bcr-Abl fusion protein. Within the BCR gene, 3 translocation breakpoints were mapped. 40 This leads to the expression of proteins with 210 kDa (termed Bcr-Abl p210), 185/190 kDa (termed Bcr-Abl p185), or rarely 230 kDa (termed Bcr-Abl p230) apparent molecular weight (Fig. 3; reviewed in Wong and Witte 41 ). These different Bcr-Abl fusion proteins are expressed in different diseases. p210 expression is the molecular hallmark of chronic myelogenous leukemia (CML). 40 The other major disease in which the Ph chromosome is detected are 20% to 30% of adult and 2% to 3% of pediatric (B-cell) acute lymphoblastic leukemias (ALLs). In Ph-positive B-ALL, around one-third of patients express p210 and around two-thirds express p185. p230 is rarely expressed in neutrophilic-CML/chronic neutrophilic leukemia. 42
Molecular Mechanisms of Oncogenic Transformation
The expression of the Bcr-Abl fusion protein has interrelated consequences. First, the Abl kinase domain becomes catalytically very active and phosphorylates a variety of different substrate proteins. Second, the Bcr-moiety of the fusion contributes critical domains and sequence motifs that drive activation of downstream signaling pathways. Third, Bcr-Abl becomes strongly autophosphorylated. Therefore, Bcr-Abl serves as a docking scaffold for SH2 and PTB (phosphotyrosine-binding) domain-containing proteins that assemble a multiprotein complex from which signaling pathways diverge. In contrast to autoinhibited Abl, Bcr-Abl was shown to exist in a complex with a limited number of other proteins in equimolar stoichiometry and different associated enzymatic activities. 43
The Coiled-Coil Domain
When comparing the primary structure of Bcr-Abl and Abl, it becomes apparent that the lack of autoinhibitory N-terminal myristoylation may contribute to the constitutive activation of Bcr-Abl (Fig. 3). In addition, activation is strongly driven by the coiled-coil oligomerization domain that is located at the N-terminus of Bcr-Abl (Fig. 3). Loss-of-function mutants have decreased kinase activity and display impaired transformation. 44 Structural and biophysical work showed that the coiled-coil domain is predominantly forming stable antiparallel homotetramers.45,46 Targeting the coiled-coil domain dimerization interface has been successfully attempted using a peptide competitor in cell lines and inhibited Bcr-Abl signaling.47,48 Despite these promising results, further investigation will have to demonstrate whether coiled-coil inhibitors work in vivo and if the required specificity for Bcr-Abl can be achieved.
Phosphotyrosine 177
The other major known contribution of the Bcr-moiety to leukemogenicity of Bcr-Abl is a tyrosine residue at amino acid position 177 (Tyr-177), which is phosphorylated (Fig. 3). Mutation of Tyr-177 to Phe strongly impaired transformation in vitro and leukemogenesis in mouse models.49,50 Phosphorylated Tyr-177 binds to the SH2 domain of the adaptor protein Grb2, which in turn binds via its 2 SH3 domains to a variety of different signaling proteins. Among those, binding and activation of the guanine nucleotide exchange factor Sos1 lead to activation of Ras and a variety of downstream effectors, including the mitogen-activated protein (MAP) kinase pathway. In addition, Grb2 binds to Gab2, which is phosphorylated strongly at multiple tyrosine residues in Bcr-Abl–positive cells. 51 The Gab (Grb2-associated binder) proteins are a family of adaptor proteins that have been shown to bind different receptor tyrosine kinases such as epidermal growth factor receptor (EGFR), c-Met, insulin receptor, and cytokine- and B-cell receptors.52,53 Homozygous Gab2 knockout cells are severely compromised in Bcr-Abl– mediated oncogenic transformation and leukemogenesis in a mouse model. 54 By means of its multiple tyrosine phosphorylation sites, Gab2 serves as an assembly platform for the tyrosine phosphatase SHP2 (PTPN11), the p85α/β regulatory subunits of PI3K, phospholipase C-γ, and other proteins without (associated) enzymatic activities. 53 Binding of these 3 proteins is critical for the activation of the MAP- and PI3-kinase pathways—2 major oncogenic pathways being activated in Bcr-Abl–positive cells.54-56 In addition, it was suggested that Gab2 (and its complex members) may coordinate the binding and activation of the transcription factor STAT5—another critical player in Bcr-Abl–dependent leukemogenesis—in the cytoplasm.57,58 Targeting of Grb2 has been attempted using an SH3 domain ligand peptidomimetic, which was able to induce apoptosis and suppress colony formation in semisolid medium in Bcr-Abl–expressing cells. 59
Crk Adaptors and STAT5
A large number of signaling pathways are activated by Bcr-Abl. In fact, there is hardly any pathway that has not been described to be influenced in one way or the other by Bcr-Abl expression.60,61 In contrast, only a few proteins appear to be critical for Bcr-Abl–dependent transformation, including Gab2 (see above), Myc, 62 and CrkL and STAT5.
The Crk family of adaptor proteins is among the dominant and best-described substrates of Abl and Bcr-Abl.63,64 In particular, CrkL binding to Bcr-Abl is necessary for oncogenic transformation, 65 whereas CrkII is not. Some of the puzzling differences between CrkL and CrkII, given their high sequence identity, were recently explained in an elegant structural analysis by distinct intramolecular interactions and accessibility of the SH3 and SH2 domains. 66 A much more detailed and expert analysis on the role of Crk protein downstream of Abl kinases is provided in other review articles published in this issue.
Another central Bcr-Abl substrate is the transcription factor STAT5, which was among the first downstream effectors shown to be activated in Bcr-Abl–expressing cells. 67 STAT5 is one the few proteins that are critical for leukemia initiation and the very few that are critical for leukemia maintenance, therefore qualifying as an attractive drug target.68-70 In addition, the contribution of STAT5 expression levels to CML progression and kinase inhibitor resistance was recently demonstrated. 71 Unexpectedly, STAT5 phosphorylation in Bcr-Abl–expressing cells is independent of the canonical upstream JAK2 kinase. Furthermore, JAK2 was not required for Bcr-Abl–induced leukemogenesis or STAT5 activation in different mouse and cellular models. 72 In addition, the hypothesis that STAT5 is a direct substrate of Bcr-Abl could be convincingly consolidated. 72 These insights question the proposed targeting of JAK2 using novel, clinically approved JAK2 tyrosine kinase inhibitors to target therapy-resistant CML. Although STAT5 is a very challenging direct drug target, as it is a transcription factor and devoid of an enzymatic domain that can be targeted readily, pimozide, a small molecule identified in a screen for inhibitors of STAT5 transcriptional activity, decreased survival of CML cells resistant to kinase inhibitors. 73
Nup214-Abl
In 7% of cases with T-cell acute lymphoblastic leukemia (T-ALL), the Nup214-Abl fusion protein is expressed. As in Bcr-Abl, only the first exon of ABL1 is missing in the Nup214-Abl fusion protein (Fig. 3). Nup214-Abl is formed by the extrachromosomal (episomal) amplification of a ~500-kb region of the long arm of chromosome 9, which fuses the majority of the NUP214 exons to ABL1. 74 Nup214-Abl localizes in multiple copies to the nuclear pore complex, and this localization is necessary for its constitutive kinase activity. 75 In mouse bone marrow transplantation models, Nup214-Abl causes a T-cell leukemia with longer latency than Bcr-Abl–induced myeloid leukemias. 75 This is in line with the observed milder deregulation of tyrosine kinase activity when compared with Bcr-Abl. Likewise, Nup214-Abl and Bcr-Abl display different in vitro and cellular sensitivities for Bcr-Abl tyrosine kinase inhibitors, some differences in substrate preference, and possibly a distinct set of protein interaction partners leading to different signaling networks. This might explain the involvement of these 2 Abl fusions in different diseases. 76 As the Bcr-Abl kinase inhibitors potently inhibited Nup214-Abl–expressing cell lines, as well as showed activity in a murine xenograft model and in primary human cells from T-ALL patients, clinical investigation in patients with NUP214-ABL1–positive T-cell malignancies is warranted. 77
Other Abl Fusions
A number of other chromosomal translocation events with ABL1 and ABL2 lead to fusions with ETV6 (Tel) (t(9;12)(q34;p13)), EML1 (t(9;14)(q34;q32)), ZMIZ1 (t(9;10)(q34;q22.3)), SFPQ (t(1;9) (p34;q34)), and RCSD1 (t(1;9)(p24;q34)) (reviewed in Cazzaniga et al. 78 ; Fig. 3). In addition, in AML cases carrying the t(1;12)(q25;p13) translocation, ETV6 is fused to ABL2. 79 Each of these ABL1 or ABL2 fusions was identified in 1 to 15 cases of T-ALL, B-ALL, AML, RAEB (refractory anemia with excess blasts), or MPN (myeloproliferative neoplasms) and therefore occurs much less frequently than Bcr-Abl or Nup214-Abl fusions. In ETV6-ABL1/2, EML1-ABL1, and ZMIZ1-ABL1, sequences starting from exon 2 of ABL1/2 are included in the fusion protein, as in Bcr-Abl and Nup214-Abl. SFPQ-ABL1 and RCSD1-ABL1 are fusions with exon 4 of ABL1, which therefore do not express the Abl SH3 and SH2 domains (Fig. 3). Most ABL fusion partners encode for one or more coiled-coil regions or a PNT domain that mediates dimerization/multimerization and drives constitutive kinase activation, in analogy to Bcr-Abl. In ETV6-Abl, Tyr-314 was found to serve as a Grb2 binding site once phosphorylated and to have equivalent functions for downstream signaling as Tyr-177 in Bcr-Abl (Fig. 3). 80
Targeting Abl Oncoproteins
The deregulated kinase activity of Bcr-Abl is necessary for the maintenance of CML. Although most of the other diseases in which Abl oncoproteins are expressed carry additional genomic lesions and are less strictly dependent on aberrant Abl kinase activity, Abl is also considered an important drug target in these diseases. Therefore, inhibition of Abl oncoprotein signaling was a rational way to target these cancers. I would like to present 3 main strategies to inhibit signaling by Abl oncoproteins, using ATP-competitive, allosteric, or Abl pathway inhibitors.
ATP-Competitive Inhibitors
The most direct approach to interfere with oncogenic Abl signaling is by using compounds that inhibit kinase activity by competing with ATP binding to the kinase domain. Due to the large number of human kinases (more than 500) and the conserved structure of the kinase domain, kinases were not considered good drug targets until the late 1990s. It was thought that specificity might be hard to achieve, and due to their important functions in normal physiology, kinase inhibitors might cause intolerable toxicity. The rapid preclinical and clinical development of the Bcr-Abl inhibitor imatinib (Gleevec) changed this dogma. 81 Together with the development of humanized monoclonal antibodies targeting the extracellular domains of oncogenic receptors, small-molecule kinase inhibitors have heralded the era of molecular targeted therapies. Today, a bit more than 10 years after Food and Drug Administration approval of imatinib for the treatment of CML, a significant fraction of new drug approvals are targeted cancer therapeutics. 82
Imatinib inhibits Abl kinase activity in the 100-nM concentration range and is remarkably specific. In addition to the Abl kinases, only a few receptor tyrosine kinases (KIT, PDFRA/B, DDR1/2) and the oxidoreductase NQO2 are inhibited. 83 Administration of imatinib leads to durable remissions in the majority of CML patients and has dramatically improved their overall survival. 84 However, the occurrence of point mutations in the Bcr-Abl kinase domain that reduces imatinib sensitivity of Bcr-Abl is the leading cause of acquired drug resistance. 85 Today, several dozens of mutations in the Abl kinase domain have been identified in patients treated with imatinib. To overcome this shortcoming, nilotinib and dasatinib were developed, which both inhibit all common imatinib resistance mutations with the exception of the T315I gatekeeper mutant. Both drugs are more potent inhibitors of Abl kinase activity.86,87 Nilotinib has a similar structure to imatinib and shares its binding mode and high specificity. In contrast, dasatinib differs from imatinib in chemical structure, binding mode, and pharmacokinetic properties.88,89 Dasatinib has a rather broad specificity and inhibits the Src, Tec, Csk, and Eph families of tyrosine kinases and several Ser-/Thr-kinases besides the kinase targets of imatinib and nilotinib.83,90,91 Both nilotinib and dasatinib are approved for the treatment of imatinib-resistant or imatinib-intolerant patients, as well as for frontline treatment of CML. Both inhibitors are well tolerated, although a similar fraction of patients suffer from more severe nonhematological toxicities that are distinct among the 2 inhibitors.92,93
A small fraction of patients develop resistance against nilotinib or dasatinib or have the T315I mutation that is pan-resistant to all approved Bcr-Abl tyrosine kinase inhibitors. Several investigational drugs were developed for these patients. 94 Recently, 2 compounds—ponatinib (AP24534) and DCC-2036—were identified that inhibited the T315I and wild-type form of Bcr-Abl potently and equally well and showed promising results in animal models with Bcr-Abl T315I.95,96 A phase 2 clinical trial of ponatinib showed promising results in patients with the T315I mutation. 97
Allosteric Targeting of the Myristate Pocket and the SH2-Kinase Interface
The N-myristoyl modification of Abl binds a deep hydrophobic pocket in the C-terminal lobe of the kinase domain, which was shown to be a major autoinhibitory mechanism (Fig. 1).18,19 Bcr-Abl is not myristoylated as it lacks the first exon of Abl, but it retains the myristate binding pocket. Therefore, it was proposed that compounds that mimic myristate binding could push the regulatory interactions toward autoinhibition. 19 As a consequence, Bcr-Abl activity would be allosterically inhibited. Furthermore, such compounds should be able to inhibit imatinib resistance, causing mutations as an alternative site is being targeted. Indeed, GNF-2, which was identified in a screen for antagonists of Bcr-Abl–dependent cell growth, bound to the myristate pocket and potently inhibited Bcr-Abl wild-type and resistant forms.98,99 A combination treatment of GNF-2 and nilotinib was shown to prolong survival in a Bcr-Abl T315I mouse model. 99 Therefore, the combination of ATP-competitive and myristate pocket inhibitors represents an innovative and rational way to overcome resistance to either agent alone (reviewed in Hantschel 23 ).
A second allosteric targeting site is the interface of the SH2 and the kinase domain in active Abl. Formation of the SH2-kinase domain interface is strictly necessary for oncogenicity, as a point mutation disrupting the interface was not able to induce CML in mice, reduced Bcr-Abl kinase activity, and failed to activate STAT5. 33 This strongly highlighted the Bcr-Abl SH2-kinase domain interface as target for therapeutic intervention. As a proof of concept, an engineered high-affinity SH2 binding protein (Abl SH2 monobody) was developed to target the Bcr-Abl SH2-kinase domain interface. This monobody inhibited Bcr-Abl kinase activity, abrogated transformation, and induced apoptosis in primary human CML cells. 33 Future work will have to demonstrate if the intramolecular domain interface that buries substantial surface area can be targeted with small molecules that could be applied in combination with ATP-competitive inhibitors to treat CML or related diseases.
Targeting Proximal Signaling/Downstream Effectors
As CML stem cells do not depend on Bcr-Abl expression for their survival and are not eradicated by current ATP-competitive inhibitors, a variety of alternative targets are being explored to target these cells. These approaches have been comprehensively covered in excellent recent review articles and therefore will not be discussed further.100-102 In addition, numerous signaling molecules in the Bcr-Abl signaling network were attempted to be inhibited with the hope to overcome resistance. A few of the best-studied examples are discussed here.
The Src kinases Lyn, Hck, and Fgr are required for Bcr-Abl–induced B-ALL in a mouse model. 103 Lyn and Hck can be overexpressed in imatinib-resistant CML patients not carrying Abl kinase domain mutations, 104 and both kinases were shown to phosphorylate the critical Tyr-177 residue in Bcr-Abl.105,106 These important insights also triggered the development of tyrosine kinase inhibitors, such as dasatinib, that simultaneously target Abl and Src kinases. Although a direct comparison of drugs targeting Abl and Src kinases (e.g., dasatinib) with a drug of similar potency only targeting Abl kinases (nilotinib) has not yet been reported in a clinical trial, a comparison of different studies with similar patient populations and end points does not seem to indicate an advantage of the additional targeting of Src kinases.
In addition, the tyrosine kinase Jak2 has been proposed as a critical target in CML and a possible kinase that phosphorylates Bcr-Abl Tyr-177 based on studies in cell lines. 107 In contrast, JAK2 was recently shown to be dispensable for Bcr-Abl–dependent leukemia initiation and maintenance in vivo. 72 In addition, in the presence of Bcr-Abl, JAK2 kinase inhibitors fail to decrease activation of STAT5. 72 Independently, combination treatments of JAK2 and Bcr-Abl inhibitors in primary cells only identified a very narrow therapeutic window, suggesting very limited therapeutic potential of JAK2-Abl kinase inhibitor combinations. 108 Considered together, these results suggest that JAK2 inhibitors might not be of therapeutic use in CML.
Finally, combinations of imatinib with drugs that target signaling pathways downstream of Bcr-Abl were tested. The Ras-MAPK pathway was targeted with Grb2 SH3, farnesyl transferase (targeting Ras), Raf, MEK, or p38 inhibitors. Likewise, the PI3K-Akt pathway was targeted with PI3K or mTOR inhibitors. Most combinations showed reasonable preclinical results, but clinical trials were not initiated or showed toxicity or lack of efficacy for many of the combinations, in comparison to the exceptional efficacy and safety of Bcr-Abl tyrosine kinase inhibitors. Furthermore, restoration of Bcr-Abl activity by resistance mutations appears to be dominant and overrides any additive or synergistic inhibitory effects of the second drug.
General Thoughts on Oncogenic Networks and Outlook
Expression of Bcr-Abl and other Abl oncoproteins leads to a qualitative and, in particular, a quantitative change of the phosphorylation state of the proteome. Bcr-Abl–expressing cell lines are rich sources for phosphoproteomics analysis and have been extensively characterized.51,109-111 Due to the variety of pathways that are activated in a Bcr-Abl kinase activity-dependent manner, some of which were described in more detail above, many tyrosine, as well as serine-threonine, kinases are constitutively activated and result in aberrant phosphorylation of numerous proteins on serine, threonine, and tyrosine residues. This fact makes it very difficult to distinguish direct Bcr-Abl substrates from those that are phosphorylated by concomitant activation of downstream kinases. One may even hypothesize that many substrates that Bcr-Abl phosphorylates would never get phosphorylated by Abl in “healthy” cells because the substrate is localized to a different subcellular site, has a suboptimal phosphorylation consensus sequence, is normally quickly dephosphorylated, or its abundance is low. One has to keep in mind that Bcr-Abl reaches activity levels for prolonged times, which have never been observed experimentally for Abl.
With a few exceptions, we know very little about the precise biological functions of the multitude of Bcr-Abl phosphorylation sites. Particular phosphorylation events may seem to make sense as they are known to support growth, proliferation, inhibition of apoptosis, or any other feature that supports or is essential for the survival of the tumor cell. On the other hand, we may be witnessing the aberrant but nonphysiological alteration of phosphorylation events by Bcr-Abl expression, which at first sight may not make biological sense. Examples include the activation of negative Abl regulators (e.g., phosphatases) or growth antagonistic and apoptosis-promoting pathways. But the balance is important, and as long as the phosphorylation event does not interfere with the overall survival of the tumor cell, this may be tolerated. Another important point to consider is that Bcr-Abl expression is a somatic pathological event and that Bcr-Abl–expressing cells did not naturally evolve. Therefore, the Bcr-Abl signaling network never had to withstand long-term selective pressure, which in physiological pathways/network evolution gets rid of dead-end or nonadvantageous interaction partners and signaling mechanisms. We have seen the emergence of new technologies to comprehensively and quantitatively measure signaling events in an unbiased way over the past decade. Together with the easier use of structural biology techniques and quick, cheap, and easy access to genomics and transcriptomics, we now have the opportunities to study cancer cell signaling across oncoproteins, diseases, tissues, and so on to decipher some of the logic (or illogicality if you wish) that underlies the aberrant signaling by kinase oncoproteins.
Finally, although great progress in treating CML patients with tyrosine kinase inhibitors has been made over the past decade, there are still pressing clinical problems. These include short-lived responses in advanced-phase CML and other diseases in which Abl oncoproteins are expressed, compound mutations (2 or more mutations in the same clone), as well as yet unidentified resistance mechanisms. Most important, none of the above-described kinase inhibitors is curing patients, as cancer stem cells are not being targeted. 112 Therefore, the current clinical guidelines suggest indefinite treatment, which is associated with problems of adherences and long-term tolerability. 113 Thus, research on Abl kinases is still a very active and exciting field of research, and the identification of alternative targeting strategies will certainly be able to overcome some of the shortcomings of current therapies.
Footnotes
Acknowledgements
I thank the ISREC Foundation for generous financial support of my laboratory.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Hantschel received speakers honoraria from Bristol-Myers Squibb and was compensated for being a member of a scientific advisory board of Novartis.
Funding
Research of the Hantschel lab is supported by the ISREC Foundation and the Swiss National Science Foundation.