
Abstract
The role of Src in tumorigenesis has been extensively studied since the work of Peyton Rous over a hundred years ago. Src is a non–receptor tyrosine kinase that plays key roles in signaling pathways controlling tumor cell growth and migration. Src regulates the activities of numerous molecules to induce cell transformation. However, transformed cells do not always migrate and realize their tumorigenic potential. They can be normalized by surrounding nontransformed cells by a process called contact normalization. Tumor cells need to override contact normalization to become malignant or metastatic. In this review, we discuss the role of Src in cell migration and contact normalization, with emphasis on Cas and Abl pathways. This paradigm illuminates several chemotherapeutic targets and may lead to the identification of new biomarkers and the development of effective anticancer treatments.
Src and Cancer
In 1911, Peyton Rous described a “transmissible agent” from cell free extracts of chicken sarcoma that could transmit tumors when injected into healthy chickens.1,2 This finding laid the foundation for the discovery of the Rous sarcoma virus and its ability to transform cells. Throughout the 1970s, many scientists carried out key genetic experiments mutating various portions of the Rous sarcoma virus genome to identify the v-Src gene (viral sarcoma gene).3-5 This gene was sequenced in the early 1980s by the groups of Bishop, Hanafusa, and Gilbert.6-8
Huebner and Todaro proposed that all animal cells contain viral “oncogenes” that are transmitted to progeny cells in a repressed form as “proto-oncogenes.”5,9,10 The cellular version of v-Src was confirmed by seminal work carried out by the groups of Varmus, Bishop, Cooper, and Hanafusa.11-14 Proto-oncogenes, exemplified by Src, can cause cancer when altered by physical or chemical agents. 9
Structurally, v-Src (viral Src) is different from c-Src (cellular Src) in that it has a number of point mutations and carboxy-terminal variations. In addition, being cellular in origin, c-Src has exons and noncoding introns that are not found in v-Src. 15 Both v-Src and c-Src encode a 60-kDa phosphoprotein with tyrosine kinase activity.16-19 However, unlike v-Src, c-Src has low transforming ability unless it is overexpressed and/or mutated. 10
As with most tumor cells, Src-transformed cells display characteristic phenotypes that include rounded morphology, increased motility, uncontrolled proliferation, anchorage independence, and growth factor independence. Furthermore, Src leads to the disruption of the actin cytoskeleton as well as cell-cell and cell-matrix adhesions.20,21 This disruption of the microenvironment and intercellular communication may lead to loss of contact growth inhibition that is also exhibited by many transformed cells.
Src Structure
Src belongs to a 9-member family of non–receptor tyrosine kinases that have overlapping biological activities. The Src family kinases (SFK) are induced by diverse families of receptors.22-24 In normal physiology, Src is implicated in pathways involving cell proliferation and differentiation, cell adhesion, migration and invasion, angiogenesis, and bone metabolism (reviewed extensively in Thomas and Brugge 23 ).
Src is composed of several functional domains21,25 that are illustrated in Figure 1. The N-terminal region of Src contains an SH4 (Src homology 4) domain and is myristoylated, which is required for association with the inner side of the cell membrane. The unique domain is relatively nonconserved and helps dictate specialized functions of each Src family kinase member. Interactions between Src and other proteins are mediated by SH2 and SH3 domains, which are regulated by phosphorylation events. In particular, the SH3 domain of Src recognizes proline residues and interacts with kinase domains and linker regions in other proteins. 26 The Src SH2 domain binds phosphorylated tyrosine on its carboxyl tail at Tyr527, as well as other proteins.27,28 The catalytic domain harbors the “activation loop” with Tyr416 phosphorylation being crucial for substrate binding. Both v-Src and c-Src are autophosphorylated at Tyr416 (Tyr419 in humans), which positively regulates its kinase activity. However, the kinase activity and transforming ability of c-Src is negatively regulated by phosphorylation at Tyr527 (Tyr530 in humans), which is absent in v-Src.21,29,30
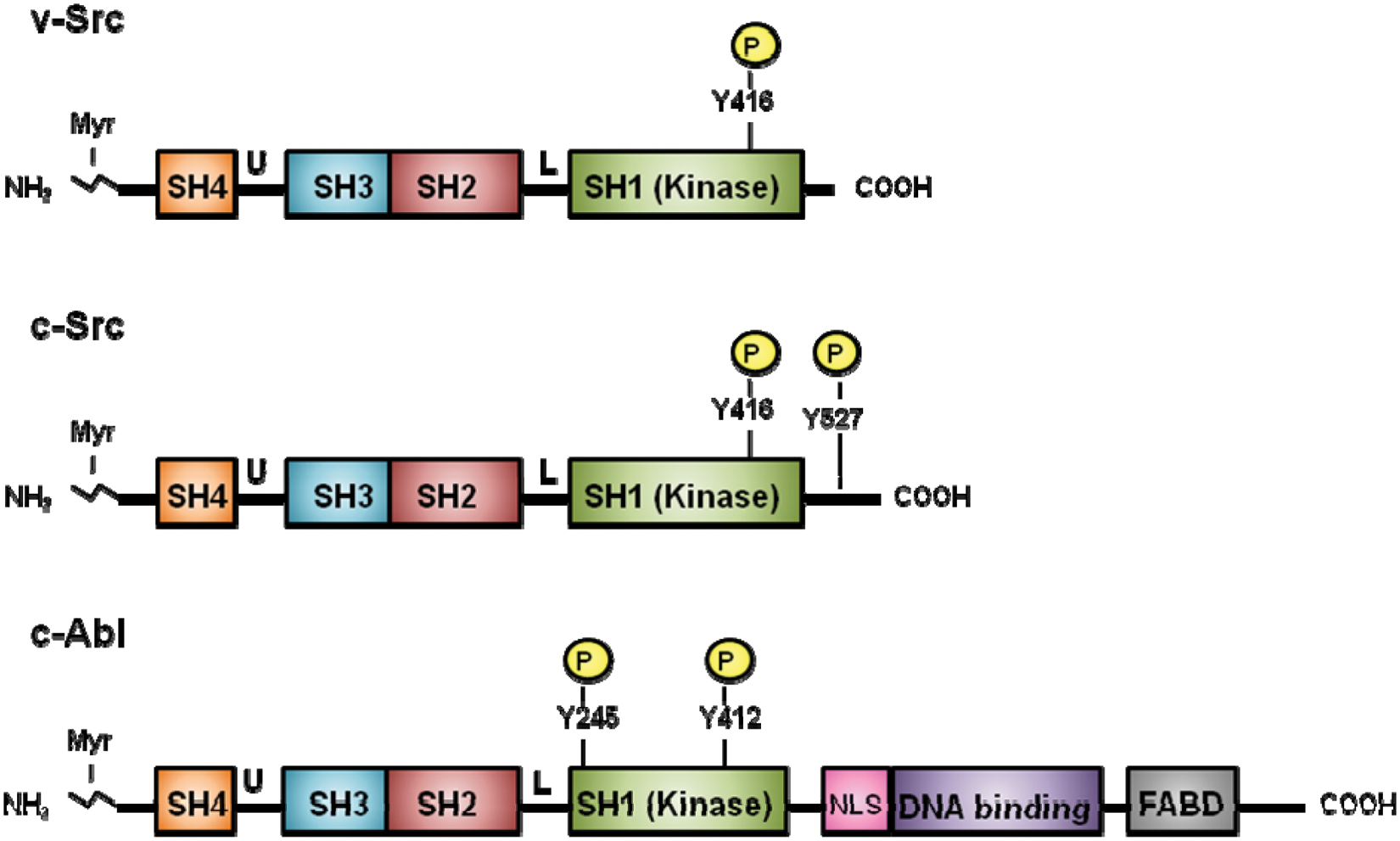
Domain architecture of v-Src, c-Src, and c-Abl proteins. The N-terminus of the proteins is myristoylated (Myr). Positions of the SH4, unique (U), SH3, SH2, linker (L), SH1 (kinase), and C-terminal domains of v-Src, c-Src, and c-Abl are shown. v-Src has a shorter, substituted C-terminal domain than c-Src. Src and Abl both contain an SH3 domain, SH2 domain, and catalytic SH1 domain in similar orientation. Specific tyrosine residues in the catalytic domains are phosphorylated when active. However, Abl has additional sequences, including a nuclear localization signal (NLS), DNA binding domain, and F-actin binding domain (FABD), which are not present in Src.
Src is clinically relevant as it plays an important role in oncogenesis. Although Src expression and its specific activity are frequently elevated in a number of human cancers, it is rarely mutated.31,32 Nonetheless, Src is a key player in the maintenance of the neoplastic phenotype and promotes cell migration and invasion, which have been linked to tumor progression and metastases.33-36 This has led to the development of several Src inhibitors as potential chemotherapeutic reagents. 35
Src and the Search for Molecular Targets
How does Src control the wide spectrum of signaling pathways that lead to oncogenic transformation? As a non–receptor tyrosine kinase, transformation clearly results from events initiated by Src phosphorylating other proteins in the cytoplasm. 5 The search for Src substrates was kicked off to elucidate this process.
Antibodies to phosphorylated tyrosine were used as tools to identify Src substrates that mediate various downstream signaling events. For example, the membrane-associated protein p36 (calpactin I/annexin II) was the first Src substrate identified in transformed cells. 5 However, it was soon apparent that Src interacts with many other proteins to elicit a multitude of cellular responses.
Src and Cas
As shown in Figure 2, upon activation by integrins, Src phosphorylates focal adhesion proteins, including Cas (Crk-associated substrate). As its name denotes, Cas associates with Crk (CT10 regulator of kinase), a linker protein containing SH2 and SH3 domains.37-39 Src, Cas, and Crk work together to regulate cell adhesion, migration, and morphological changes associated with neoplastic transformation.23,40,41 After immunoaffinity purification of Cas from cells expressing v-Crk, Sakai et al. 42 isolated the cDNA for Cas and confirmed that it served as an efficient Src kinase substrate.
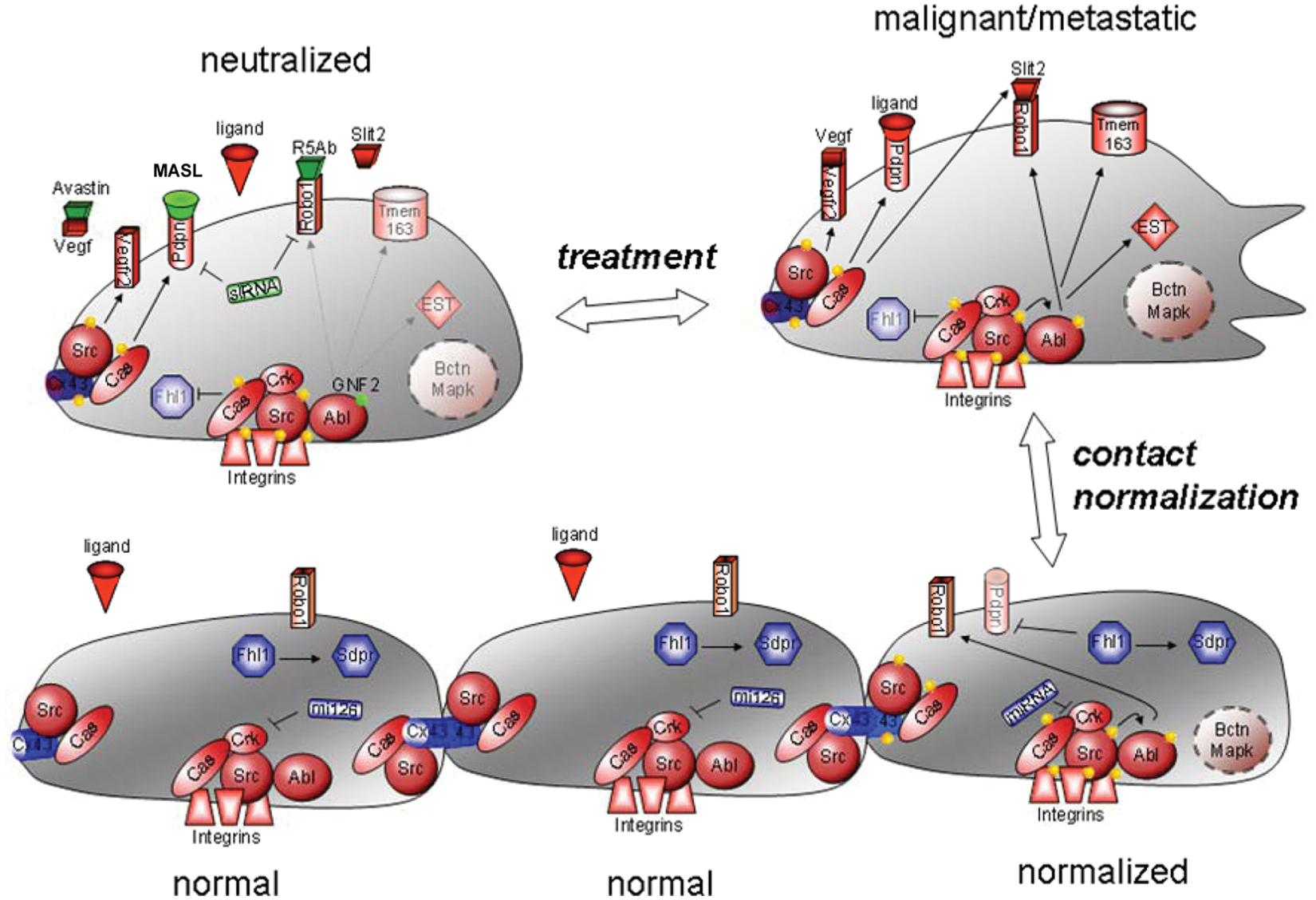
Contact normalization and neutralization of tumor cell growth. Cancer cells can be normalized by adjacent nontransformed cells. Src phosphorylates Cas, which associates with Cx43 and Crk to block communication between cells110,136 and inhibit Fhl1 and Sdpr expression to promote nonanchored cell growth and motility.30,33,112,134,135 Src also activates Abl to stabilize Robo1, which, upon stimulation by Slit2, activates Rho GTPases, which recruit downstream effectors such as N-WASP to promote cell migration. 75 Other receptors,67,134 adaptor proteins,2,30,82,135,136 and molecules, including miRNAs,81,82 that promote or inhibit this process are indicated in red and blue, respectively. These molecules can be used as biomarkers to detect cancer, as well as targets to neutralize cancer cells. For example, kinase blockers, 67 siRNA,67,82,134 antibodies, 67 and receptor blockers, including MASL (green), can target proteins involved in contact normalization to suppress cancer progression downstream of Src kinase activity. 166
The domain architecture of Cas (and of the related family members EFS/Sin and HEF1) is consistent with its role as an adaptor protein and a central hub of signaling.43,44 As shown in Figure 3, the N-terminus of Cas contains an SH3 domain that binds to focal adhesion kinase45,46 and other proteins containing polyproline motifs.43,44 Following a proline-rich sequence, the large central portion of Cas (the “substrate domain”) contains multiple YXXP sequences that are phosphorylated by Src and other tyrosine kinases (described in more detail below). On the C-terminal side of the substrate domain lies a serine-rich domain that forms a 4-helix bundle and interacts with 14-3-3 proteins.47,48 At the C-terminus of Cas, Tyr668 (in the sequence pYDYV) fits the consensus sequence for high-affinity Src SH2 domain binding. Both Tyr668 and the C-terminal polyproline sequence of Cas are known to be binding sites for Src,49-52 and mutations in this region result in impaired phosphorylation of Cas in vivo. Other kinases and signaling molecules interact with the C-terminal domain of Cas and related proteins.43,44
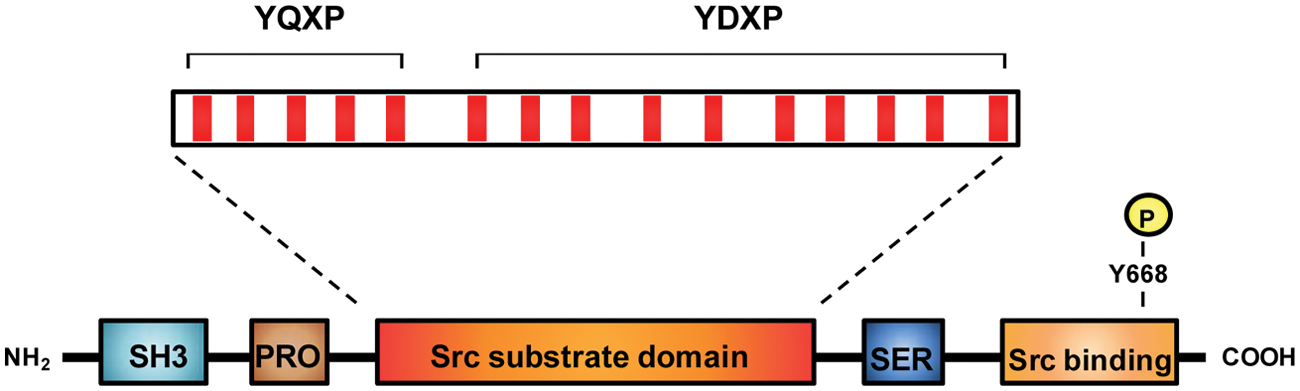
Domain architecture of Cas. Cas contains an SH3 domain, proline-rich sequences (Pro), a substrate region containing 15 YXXP repeats (red), a serine-rich region (Ser), and a C-terminal Src binding site. The Src binding site contains a proline-rich sequence that binds to the SH3 domain of Src (residues 639-647: RPLPSPPKF) and a phosphorylated Tyr (Y668) that binds to the SH2 domain of Src.
Numerous studies have focused on the biological roles of Cas in normal cell physiology, as well as the importance of Cas in Src-mediated cellular transformation (reviewed more extensively in Bouton et al., 43 Tikhmyanova et al., 44 Chodniewicz and Klemke, 53 and Defilippi et al. 54 ). Cas undergoes rapid phosphorylation following mitogenic stimulation and in response to fibronectin attachment via integrin receptors.53,55-57 It has been implicated in processes as diverse as cell migration, cell cycle control, apoptosis, and signal transduction by a variety of transmembrane receptors.43,58 Mouse embryos lacking Cas die in utero due to defects in actin stress fiber and myofibril organization, impaired cardiovascular development, and growth retardation. 59 Fibroblasts derived from these Cas-deficient mice contain disorganized and shortened actin stress fibers, 60 consistent with a role for Cas in the regulation of the actin cytoskeleton. Cas-deficient fibroblasts are also partially resistant to transformation by Src, suggesting that Cas represents an important cellular substrate in the conversion to the transformed phenotype. 59
Cas plays a key role as an adaptor protein to promote cell migration. As mentioned above, Cas is concentrated at focal adhesions,45,46,61 and it is important in regulating the turnover of these structures. Tyrosine phosphorylation of Cas is increased upon engagement of integrins by extracellular matrix components such as fibronectin, vitronectin, and laminin.45,46,62 The intermolecular interaction between Cas and Crk is believed to be a critical component of this response. As described above, the central substrate domain of Cas contains about 15 potential phosphorylation sites with the consensus YXXP. Nine of these motifs are potential Crk SH2 domain-binding sites, 63 and integrin-dependent activation of Cas results in the formation of Cas-Crk complexes.64,65
The formation of Cas-Crk complexes has been described as a “molecular switch” for the induction of cell migration. 64 Overexpression of Cas promotes the migration of human pancreatic carcinoma cells plated on vitronectin, but mutant forms of Cas lacking the substrate domain are not able to drive migration. Expression of an active form of the Abl tyrosine kinase disrupts the Cas-Crk complex and prevents cell migration. 66 A dominant negative form of the small GTPase Rac1 blocks migration, suggesting that Rac1 lies downstream of the Cas-Crk complex. Rac1 is thought to be activated by recruitment of the guanine nucleotide exchange factors C3G and DOCK180 to the complex.67,68 In addition to integrin-dependent responses, this signaling pathway also operates to facilitate migration in response to growth factors.64,69
Several studies have focused on the mechanism and specificity of Cas tyrosine phosphorylation within the substrate domain. The YXXP motifs within the substrate domain can be divided into an N-terminal group of YQXP motifs and a C-terminal group of YDXP motifs (Fig. 3). Deletion studies provide evidence that the YDXP motifs play a particularly important role in actin cytoskeletal organization and cell migration. 70 Variants of Cas that contain only 4 or 5 of the YDXP tyrosines are functional in migration assays, although with reduced potency compared with wild-type Cas. 71 Mutation of the tyrosine in one particular YDXP site (Y253) decreases migration in response to Src expression but shows no effect on the ability of Src to increase growth rate or promote anchorage independence. 40
Kinetic experiments show that Src phosphorylates the substrate domain of Cas via a processive mechanism, in which Src binds to the C-terminus of Cas and phosphorylates all available sites prior to dissociating. 52 Mutation of individual YXXP sites shows that individual sites are not required for processive phosphorylation and that there is no defined temporal sequence of phosphorylation. 72 The processive mechanism leads to a high rate of multisite phosphorylation of Cas by Src and faster assembly of Cas-Crk complexes. Phosphorylation of the substrate domain YXXP motifs has also been demonstrated to be important in primary tumor growth and metastasis in an in vivo lung metastasis model. 73
Src and Abl
In addition to Cas and other adaptor proteins, Src phosphorylates specific protein kinases to augment mitogenic and transforming signaling cascades. For example, Src phosphorylates Abl to activate Rho GTPases and reorganize the actin cytoskeleton to promote cell migration.74,75 Like Src, Abl is a non–receptor tyrosine kinase that has been implicated in several human cancers, including leukemia, breast, and lung cancer. 76 The clinical relevance of Src and Abl is evidenced by several pharmacological kinase inhibitors currently used as chemotherapeutic medicines.77-80
As shown in Figure 1, Src and Abl are both myristoylated, which drives them to the plasma membrane.74,76,81 Src and Abl both contain an SH3, SH2, and catalytic (SH1) domain in similar orientation. As with Src, Abl SH2 domains associate with specific peptide sequences containing phosphorylated tyrosine residues,27,28 whereas SH3 domains associate with proline-rich motifs in binding partners. 26 Phosphorylation of specific tyrosine residues in the catalytic domains of both proteins occurs when the enzymes are in an active conformation.76,81 Autoinhibition of Src kinase activity is mediated by interaction of the Src SH2 domain with its phosphorylated (Tyr527) C terminus. In contrast, autoinhibition of Abl kinase activity is brought about by binding of the N-terminal myristoylated site with its kinase domain in a conformation similar to inactive Src. 82 The Abl kinase can be activated by Src family kinases, including Src and Fyn.74,83 Src activates Abl by phosphorylating Y412 in the activation loop of the catalytic (SH1) domain. 74
Although both are non–receptor tyrosine kinases with similar architecture, Abl and Src are quite distinct from each other. Abl is about twice the size of Src and contains additional regions, including actin and DNA binding domains in the carboxyl part of the protein (see Fig. 1). In contrast to the oncogenic effects of Abl kinase activity at the plasma membrane, Abl also interacts with DNA in the nucleus to inhibit cell proliferation. 84 The nuclear localization and DNA binding motifs needed for this antiproliferative effect are not present in v-Abl, which can promote tumor cell growth and motility.85,86
Like Src, the Abl kinase can phosphorylate a plethora of proteins that are involved in the regulation of cell migration and anchorage dependence, including the integrin adaptor proteins Crk and Cas. 87 Abl also associates with the Rho family of GTPases that modify effectors, including N-WASP, to regulate the actin cytoskeleton and microtubule dynamics that drive cell migration as described below.83,88,89
Src and Robo1
A number of growth factors, including epidermal growth factor (EGF) and platelet-derived growth factor (PDGF), activate Src, which, subsequently, activates the Abl kinase to promote cell migration. In addition, Src can directly activate Abl to promote migration without the need for growth factor stimulation. This is confirmed in that Abl blockers can also suppress the migration of growth factor–stimulated nontransformed cells or Src-transformed cells.74,83 Thus, Src activates Abl to promote tumor cell migration and invasion.
Results from nonbiased microarray experiments indicate that Src can increase or decrease the expression of many genes—possibly over 8% of the entire transcriptome.90,91 Some of these genes encode factors that promote cell migration. For example, Src induces the expression of Slit2. 90 Slit2 is a secreted protein that binds to a receptor called “Robo1.”92,93
Robo1 is a transmembrane receptor of the immunoglobulin family.89,94 Upon Slit2 binding, Robo1 works with the Abl kinase to rearrange the actin cytoskeleton and induce cell migration.92,95 Indeed, cell migration can be inhibited by blocking Robo1 receptor activity with a monoclonal antibody to the extracellular domain of the protein. 92
Studies have implicated Robo1 in cancer of the liver, breast, and brain.96-99 Moreover, Robo1 signaling can augment tumor angiogenesis.93,95 Indeed, as illustrated in Figure 2, Src activates Abl, which, in turn, stabilizes Robo1 to promote tumor cell migration. 75
Cell migration is required for a myriad of events, including development, wound healing, tumor cell invasion, and metastasis. Ultimately, the actin cytoskeleton must be altered for cells to migrate.100,101 Rho GTPases play a critical role in this process.102,103
Rho GTPases are members of the Ras superfamily. There are over 20 mammalian Rho GTPases. When bound to GTP, these GTPases target effectors to modify the actin cytoskeleton. 104 In particular, 3 of the Rho GTPases—Rho, Rac, and Cdc42—activate members of the Wiskott-Aldrich syndrome proteins (WASPs) and WASP verprolin homologous proteins (WAVEs), which, in turn, activate actin polymerization factors, including Arp2/3 and formin, to cause elongation and branching of actin fibers.101,103,105
Many axon guidance receptors, including Robo1, regulate the Rho family of GTPases to effect changes in motility. For example, N-WASP is an effector through which Rho GTPases regulate the actin cytoskeleton. 89 As shown in Figure 2, studies indicate that Src activates Abl to stabilize Robo1 at the plasma membrane where it activates Rho GTPases and N-WASP to promote transformed cell migration. 75
Contact Normalization
Nontransformed cells can force tumor cells to assume a normal morphology and phenotype.90,106,107 This observation was reported over 50 years ago.108,109 In vivo, genetically transformed cells can assume a normal phenotype and reside as “occult tumors” in a variety of tissues, including skin and mammary epithelium.110,111 Cells transformed by chemicals, tumor viruses, and oncogenes such as Src can be normalized by contact with nontransformed cells.90,112-114 This suppression of the neoplastic phenotype by direct contact between transformed and nontransformed cells is called contact normalization.
Contact normalization is mediated by intercellular junctions, potentially adherens and gap junctions.106,107 Both are targets of Src phosphorylation. Src can phosphorylate cadherins and β-catenin to disrupt adherens junctions.115-117 A characteristic feature of neoplastic cells is the epithelial-mesenchymal transition (EMT) wherein acquisition of invasive capabilities is associated with a decrease in E-cadherin and an increase in N-cadherin expression. 118
In addition to cadherins, Src can phosphorylate connexins, particularly Cx43, to disrupt gap junctional communication between cells.91,119 In general, most cancer cells show reduced gap junctional communication compared with their nontransformed precursors. In addition, forced Cx43 expression can suppress tumor cell growth and migration. The ability of nontransformed cells to regulate the growth of adjacent tumor cells has been correlated with Cx43 expression and gap junctional communication between these cell types.120-123 However, studies indicate that gap junctions are not required for contact normalization.90,106 Nonetheless they may augment the ability of normal cells to control the growth of neighboring tumor cells.121,124-126
The presence of several adhesion molecules, including connexins and cadherins, on the cell surface may help maintain normal cell phenotypes and safeguard against malignancies. 106 Src phosphorylates both of these proteins to disrupt these intercellular communication signals. Indeed, although Src does not require Cas to phosphorylate Cx43, Src does require Cas to block gap junctional communication between transformed cells.91,127 Thus, Cas appears to act as a switch between intercellular communication and tumor cell migration downstream of Src kinase activity.
Tumor cell motility is clearly one of the most dangerous properties manifested by aggressive malignancies.128,129 Understanding how this property is controlled by contact normalization would shed light on mechanisms that underlie invasive and metastatic tumorigenesis. As described above, Src phosphorylates specific sites on Cas, which acts as a molecular switch driving fundamental hallmarks of tumor cell growth and migration. Evaluation of proteins enhanced by Src-Cas signaling and reversed by contact normalization can be used to identify functionally relevant cancer biomarkers and chemotherapeutic targets.
Src, Contact Normalization, and Novel Tumor Suppressors
Comprehensive analysis of gene expression indicates that transforming Src activity affects the expression of over 8% of the transcriptome, and less than 0.01% of these genes are inversely affected by contact normalization.90,91 By this approach, genes repressed during transformation but induced during contact normalization are likely to act as tumor suppressors. Four-and-a-half LIM domains 1 (Fhl1) and serum deprivation response protein (Sdpr) are examples of tumor suppressors that are downregulated by Src-Cas signaling and induced during contact normalization to inhibit tumor cell migration and nonanchored growth. 41
The Fhl1 protein contains an N-terminal half LIM domain followed by 4 complete LIM domains (cysteine-rich zinc finger motifs named after their discovery in Lin11, Isl-1, and Mec-3). Fhl1 can move between intercellular junctions, focal adhesions, and the nucleus to affect gene expression 41 and cellular processes, including proliferation, differentiation, apoptosis, adhesion, migration, transcription, and signal transduction.130,131 For example, Fhl1 can induce expression of Sdpr, 132 which is a phosphatidylserine-binding protein that is induced during growth arrest by serum deprivation of nontransformed cells but not transformed cells. 107
Forced Fhl1 expression (e.g., by transfection) inhibits anchorage-independent growth and migration of Src transformed cells. As illustrated in Figure 2, Src uses Cas to block gap junctional communication and inhibit Fhl1 expression. These events are reversed by contact normalization, 41 which appears to be independent of MAPK activity. 132 In addition to inducing Sdpr production, reduced Fhl1 expression correlates with reduced p21 and increased c-Myc levels. Also, a decrease in cancer cell growth depends on the interaction of Fhl1 with CK1δ (casein kinase 1δ) and Smad4 with a concomitant increase in Smad2/3 phosphorylation. 130
Fhl1 downregulation has been observed in several human cancers, including astrocytoma, mammary carcinoma, renal carcinoma, prostatic carcinoma, melanoma, hepatocarcinoma, pulmonary adenocarcinoma, and gastric cancer.41,130,132-135 Accordingly, low expression of Fhl1 correlates with deep tumor invasion of the serosal layer in patients with primary gastric cancer. 133 Taken together, these data indicate that Src uses Cas to inhibit Fhl1 expression to promote tumor cell growth and migration, and this process is reversed by contact normalization.
Src, Contact Normalization, and miRNA Expression
MicroRNAs (miRNAs) are a class of small noncoding RNAs of 18 to 25 nucleotides in length that inhibit mRNA translation or induce mRNA degradation to regulate posttranscriptional gene expression. miRNAs play a critical role in a wide range of biological processes such as cellular proliferation, embryonic development, and tumorigenesis. 136 A direct link between miRNAs and cancer was first recognized when miR-15 and miR-16 were deleted or downregulated in B-cell chronic lympholytic leukemia (B-CLL). 136
Studies have revealed that miRNAs can function as tumor promoters or suppressors that modulate cancer-related genes involved in cell proliferation and apoptosis. 137 Also, there is growing evidence linking miRNAs to tumor cell migration and metastasis. For example, inhibition of miR-126 promotes lung cancer migration and invasion, 138 whereas upregulation of miR-224 increases hepatocellular carcinoma cell proliferation, cell migration, and invasion. 139
miRNAs are regulated by Src transformation and contact normalization. Some experiments indicate that the expression of approximately 10% of oncomirs (miRNAs related to oncogenesis) is significantly affected by Src kinase activity and that this effect is reversed in about 1% of these. For example, miR-126 is suppressed several fold in Src-transformed cells compared with nontransformed cells but induced in transformed cells undergoing contact normalization. In contrast, miR-218 and miR-224 are induced by Src but not affected by contact normalization. 140
Interestingly, miR-126 targets Crk, which is phosphorylated by Src and associates with Cas to promote tumor cell migration and invasion as discussed above. 141 Crk is frequently deregulated in a number of aggressive human tumors, including that of lung, breast, glioblastoma, and sarcomas. 142 As shown in Figure 2, Src suppresses miR-126 expression to augment Crk in order to promote tumor cell migration, and this effect can be reversed by increasing the expression of miR-126. 140
In contrast to miR-126, Src transformation increases miR-218 and mi4-224 expression. miR-224 promotes nonanchored growth of Src-transformed cells, which is consistent with its elevated expression in several tumors. 140 Furthermore, Src downregulates the expression of oncomirs, including miR-99a and miR-542-3p, to promote tumor progression.143,144 Further elucidation of miRNA pathways should elucidate their clinical relevance and use as biomarkers and potential chemotherapeutic targets.
Src and Downstream Chemotherapeutic Targets
As discussed above, Src can downregulate tumor suppressors that are induced by contact normalization (exemplified by Fhl1 and miR-126). Contrapositively, Src induces the expression of specific tumor promoters that are downregulated during contact normalization. As expected from effectors of the Src-Cas signaling pathway, these tumor promoters tend to augment cell motility. As illustrated in Figure 2, these proteins include the extracellular receptors Tmem163, Vegfr2, and Pdpn. 127 It should be stressed that these genes were found to be the most affected by contact normalization or Src-transformed cells from over 30,000 genes represented by about 45,000 probe sets. 127 These receptors can serve as functionally relevant cancer biomarkers and chemotherapeutic targets.
Tmem163 (transmembrane protein 163) is predicted to form an integral membrane protein with 6 transmembrane helices. Although recently discovered, experiments indicate that Src induces Tmem163 expression in order to promote cell migration. 127 Increased Tmem163 mRNA expression has also been found in papillary thyroid carcinoma 145 and nodular lymphocyte-predominant Hodgkin lymphoma. 146
Vegfr2/Kdr (vascular endothelial growth factor receptor 2/kinase-insert domain receptor) is a receptor tyrosine kinase that plays an important role in tumor growth, invasion, and angiogenesis. 147 Vascular endothelial growth factor (VEGF) ligands interact with Vegfr2 to trigger its kinase activity. VEGF antibodies and other inhibitors can be used to block Vegfr2 signaling and downstream signaling events.147,148 For instance, the antibody bevacizumab (Avastin) targets VEGF-A to inhibit angiogenesis in metastatic cancers. 149 In addition, soluble decoy receptors can act as “VEGF-Traps” to suppress tumor growth and vascularization. 150 Kinase blockers, including sunitinib malate (Sutent), can directly target Vegfr2 to inhibit its kinase activity.147,151
Podoplanin (Pdpn, also called T1α, PA2.26, aggrus, and gp36) is a transmembrane mucin-like protein that augments tumor cell invasion. Pdpn associates with ezrin and regulates the activities of Rho GTPases to promote filopodia formation, cell motility, and metastasis.152-154 Pdpn expression is induced by tumor promoters, including TPA, RAS, and Src,127,155,156 and enhances transformed cell motility and invasion.152,157 PDPN is predominantly found at the invasive front of many tumors, which is consistent with its role in promoting malignant invasion.152,158 For example, PDPN expression is strongly induced in about 40% of breast cancers,158,159 over 50% of oral cancers,160,161 and about 80% of skin cancers.162,163 Antibodies against PDPN and a PDPN interacting partner (tetraspanin CD9) can inhibit lung metastasis of CHO cells transfected with PDPN.164,165 We have also found that specific lectins can target Pdpn to inhibit tumor cell growth and migration in vitro and in vivo. 166
Summary
The story of Src presents a seminal example of bench to bedside research. Although this story has taken over a century to unfold, the spirit of discovery infused with technological advancements is clearly leading to the development of innovative and effective ways to target the molecular causes of cancer. For example, Src inhibitors, including dasatinib, bosutinib, and saracatinib, are being used alone and in conjunction with other agents to combat cancer. Unfortunately, however, this approach is fraught with significant challenges. Src inhibitors are relatively nonspecific and may target other kinases, including c-Kit, c-FMS, EphA2, and PDGFR,35,167 to cause a variety of medical side effects. 168 Thus, in addition to Src, its downstream effectors may serve as useful targets for cancer therapy. Elucidating the Src-Cas signaling pathway and how it is affected by contact normalization should aid in the identification of functionally relevant cancer biomarkers and promising targets for cancer therapy.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported in part by grants from the Research Foundation of UMDNJ, the Northarvest Bean Growers Association, and United States National Institutes of Health RO1CA88805 to GSG and RO1CA58530 to TWM.