
Abstract
Although c-Abl and Arg non–receptor tyrosine kinases are well known for driving leukemia development, their role in solid tumors has not been appreciated until recently. Accumulating evidence now indicates that c-Abl and/or Arg are activated in some solid tumor cell lines via unique mechanisms that do not involve gene mutation/translocation, and c-Abl/Arg activation promotes matrix degradation, invasion, proliferation, tumorigenesis, and/or metastasis, depending on the tumor type. However, some data suggest that c-Abl also may suppress invasion, proliferation, and tumorigenesis in certain cell contexts. Thus, c-Abl/Arg may serve as molecular switches that suppress proliferation and invasion in response to some stimuli (e.g., ephrins) or when inactive/regulated, or as promote invasion and proliferation in response to other signals (e.g., activated growth factor receptors, loss of inhibitor expression), which induce sustained activation. Clearly, more data are required to determine the extent and prevalence of c-Abl/Arg activation in primary tumors and during progression, and additional animal studies are needed to substantiate in vitro findings. Furthermore, c-Abl/Arg inhibitors have been used in numerous solid tumor clinical trials; however, none of these trials were restricted to patients whose tumors expressed highly activated c-Abl/Arg (targeted trial). Targeted trials are critical for determining whether c-Abl/Arg inhibitors can be effective treatment options for patients whose tumors are driven by c-Abl/Arg.
Introduction
c-Abl/Arg Regulation
The ubiquitously expressed Abl family of non–receptor tyrosine kinases, c-Abl and Arg (Abl-Related-Gene), is encoded by Abl1 and Abl2 genes, respectively. 1 c-Abl and Arg N-termini are highly conserved and contain an N-terminal cap, myristoylation site, which targets the proteins to the plasma membrane, SH3, SH2, and tyrosine kinase domains. The C-termini are less conserved as c-Abl but not Arg contains nuclear localization and export signals and a DNA-binding domain, whereas both proteins have F-actin binding domains. 2 Thus, c-Abl and Arg are located at the plasma membrane and cytoplasm, while c-Abl also is observed in the nucleus. c-Abl/Arg activities are strongly inhibited by intramolecular interactions,3,4 binding cellular inhibitors (e.g., PRDX-1/PAG, AAP1, Abi1, lipids, and F-actin),5-11 dephosphorylation,12-14 and phosphorylation (Y272) 15 and are degraded via proteosomal or caspase-mediated pathways.16-18 Thus, c-Abl and Arg are activated by mutation, dimerization, phosphorylation, or binding proteins that disrupt intramolecular interactions,19-21 as well as by phosphorylation within kinase and interlinker regions (Y412, Y245).22-25 Disruption of autoinhibition, such as by translocation of Abl1 or Abl2 next to a variety of different genes (e.g., BCR, Tel, ETV6), causes constitutive activation, which drives leukemia development.26,27 Imatinib mesylate (Gleevec, STI571), an inhibitor of c-Abl, Arg, and BCR-Abl, is approved by the Food and Drug Administration (FDA) for treating BCR-Abl+ leukemias. 28 Nilotinib, a more sensitive second-generation inhibitor, shows even greater promise in inducing remission. 29 Imatinib and nilotinib also inhibit c-Kit and PDGFR, and nilotinib also targets DDR and CSF-1R.29,30 Drugs targeting Src family kinases (SFKs) and Abl kinases (SKI-606-bosutinib, dasatinib) also are used to treat BCR-Abl+ diseases, and additional second-generation drugs are in development.31,32
In nontransformed fibroblasts, c-Abl and Arg are transiently activated downstream of receptor tyrosine kinases (RTKs), PDGFR, and EGFR22,33-35 and are activated by bFGFR in endothelial cells. 36 c-Abl also is activated by transforming growth factor–β (TGF-β)37,38 and AT-1 (angiotensin subtype 1) receptors. 39 PDGFR stimulates c-Abl/Arg activation via Src family kinases (SFKs), which directly phosphorylate c-Abl/Arg on Y245 and Y412, activating the kinases,22,33 and via activation of PLC-γ, which hydrolyzes the c-Abl/Arg inhibitor, PIP2. 6 In contrast, activation of c-Abl by TGF-β involves PI3K and PAK2, while SFKs mediate c-Abl activation by AT-1. 39
Biological Function of c-Abl/Arg in Nontransformed Cells
Constitutively active forms of Abl (BCR-Abl, v-Abl) transform rat cells, whereas overexpression of wild-type c-Abl induces cell cycle arrest.40-42 c-Abl shuttles between the cytoplasm and nucleus,43,44 and acetylation and/or binding to 14-3-3 proteins promotes its cytoplasmic retention.45-47 Activation of nuclear c-Abl by DNA-damaging agents induces G1 arrest and/or apoptosis,48-54 whereas activation of the membrane and cytoplasmic pools of c-Abl and Arg by growth factors promotes membrane ruffling and motility of fibroblasts and endothelial cells, as well as endothelial tubule formation.6,10,22,33,36,55-61 In contrast, inhibition or knockout of c-Abl promotes wound-healing motility and migration toward collagen, fibronectin, or insulin.62,63 c-Abl kinase activity decreases following detachment of fibroblasts from the extracellular matrix, and adhesion to fibronectin transiently increases c-Abl activity; induces its export from the nucleus to focal contacts, 64 which prevents cell spreading and promotes the formation of F-actin microspikes and filopodia.65-67 In contrast, Arg promotes lamellipodia 68 and actomyosin contraction, as well as regulates focal adhesion size, distribution, and dynamics by binding and crosslinking microtubules and F-actin following adhesion to fibronectin.69,70 In addition to affecting fibroblast cell-matrix attachment, c-Abl/Arg also regulate epithelial cell-cell adhesion. 71 Loss of Arg expression prevents epithelial adherens junction formation and impairs the establishment of 3D polarized cysts, while expression of constitutively active Arg produces aberrant cysts with inverted polarity, indicating that Arg function not only is required for polarity but also promotes aberrant polarity in cells expressing sustained, activated Arg.71,72
Mechanisms of c-Abl and Arg Activation in Solid Tumors
Despite a critical involvement of c-Abl and Arg in the development of a variety of human leukemias, mutations and/or activating translocations have not been identified in solid tumors, thus leading to the assumption that c-Abl and Arg are not activated in these cancers. However, some early data suggested that c-Abl and/or Arg may be overexpressed in some solid tumors. c-Abl and/or Arg expression was significantly increased (assessed by immunohistochemistry [IHC]) in brain, lung, ovarian, colorectal, and prostate cancers, as well as in chondrosarcomas, liposarcomas, diffuse gastric adenocarcinomas, oral squamous carcinomas, atypical teratoid and malignant rhabdoid tumors, and endometrial carcinomas as compared with normal tissue or benign tumors, and c-Abl amplification was noted in renal medullary carcinomas.73-82 Moreover, c-Abl expression correlated with EGFR, IGF-1R, PDGFR, and/or c-Kit expression in colon cancer, lung cancer, lymphomas, and melanomas. 79 Furthermore, c-Abl and/or Arg expression positively correlated with disease progression in ovarian, 76 colon, 74 and gastric cancers, 77 whereas c-Abl expression was decreased in high-grade as opposed to low-grade chondrosarcomas and ovarian serous carcinomas.75,78 Although interesting, one has to be cautious in interpreting these IHC studies, as some c-Abl antibodies cross-react with other proteins; thus, knockout cells ideally should be used to demonstrate antibody specificity. 22 In addition, since c-Abl and Arg are tightly regulated, it is not clear whether increased expression translates to high activity. High-level c-Abl expression (>200 fold) induces constitutive activation, likely by titrating out cellular inhibitors; however, it is unclear whether the increased expression observed in solid tumors is sufficient to induce activation.6,9,10,22 Although IHC using a phosphospecific c-Abl antibody has been attempted, 80 all commercially available phospho-Abl antibodies cross-react with PDGFR and/or EGFR, and thus, the staining obtained is unlikely to reflect c-Abl activity. Lack of specific phospho-antibodies has hampered efforts to assess c-Abl/Arg activities in primary solid tumors.
Since c-Abl and Arg are transiently activated by RTKs and SFKs, we reasoned that constitutively active RTKs and/or SFKs, which are often expressed in solid tumors, may induce sustained activation of c-Abl/Arg. We observed that c-Abl/Arg expression was increased in breast cancer cell lines relative to human mammary epithelial cells and in melanoma cell lines relative to primary melanocytes.83,84 In breast cancer cells, c-Abl and/or Arg were only highly active in some lines, and activation did not correlate with expression, whereas in melanoma cells, lines with higher expression had higher activities.83,84 c-Abl/Arg activities were highest in triple-negative (ER−, PR−, Her-2−; BT-549, MDA-MB-231, MDA-MB-468) and Her-2+ (BT-474, UACC-893) breast cancer cell lines as compared with an ER+/PR+/Her-2− breast cancer cell line (MCF-7), and active c-Abl was localized in the cytoplasm, similar to BCR-Abl and v-Abl.84,85 Significantly, c-Abl/Arg activities (assessed by phosphorylation of substrates, Crk/CrkL) were dramatically elevated in primary melanomas relative to benign nevi or normal skin.83,79 We showed that constitutively active IGF-1R, Her-2, EGFR, and/or SFKs contributed to c-Abl/Arg activation in melanoma and breast cancer lines, ruling out mutation as the method of activation and uncovering a novel activation mechanism in solid tumors (Fig. 1). However, other events also are likely to contribute to c-Abl/Arg activation since RTK/SFK inhibition did not completely block c-Abl/Arg activities. 84 Sirvent and colleagues 86 confirmed our findings by demonstrating that constitutively active SFKs induce sustained activation of c-Abl in breast cancer lines. In addition, Zhao and colleagues 87 showed that c-Abl was highly expressed in 54% of primary breast cancers, particularly in ER+, late-stage cancers with lymph node involvement; however, c-Abl activity was not evaluated. Constitutively active receptor tyrosine kinases also contribute to c-Abl/Arg activation in human glioblastoma cells (PDGF) 88 and gastric and hepatocarcinoma cells (c-Met) (Fig. 1). 89 In liver cancer cells, c-Abl is activated downstream of overexpressed claudin-1, a tight junction protein whose increased expression is associated with an advanced, aggressive phenotype.90,91 High-level c-Abl expression also was observed in primary anaplastic thyroid carcinomas but not in follicular or papillary carcinomas or in normal thyroid tissue, and c-Abl expression correlated with mutant p53 expression. 91
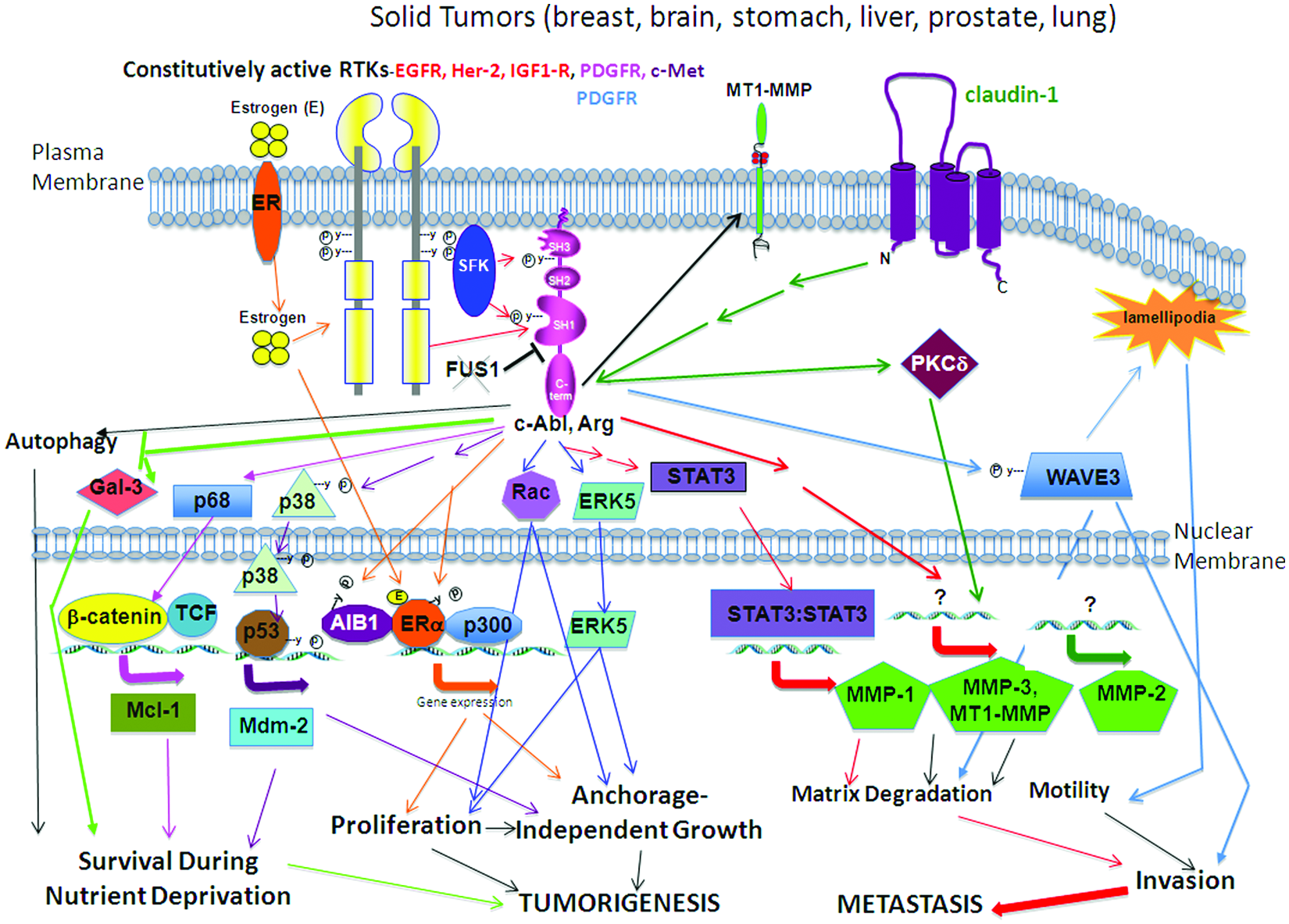
Activation of c-Abl/Arg in solid tumors. Summary of the signals that induce constitutive activation of cell surface receptors (receptor tyrosine kinases, claudin-1), in a variety of solid tumors, induces constitutive activation of c-Abl/Arg. c-Abl/Arg, in-turn, activate a variety of intracellular signaling pathways, which result in increased survival, proliferation, anchorage-independent growth, migration, matrix degradation, and invasion, processes necessary for tumor growth and/or metastatic progression. Arrow colors indicate proteins that are in the same signaling pathways.
Interestingly, Lin and colleagues 92 reported another novel mechanism of c-Abl activation in human non–small cell lung cancer (NSCLC) cell lines. They showed that the FUS1 tumor suppressor is an inhibitor of c-Abl, and loss of FUS1 expression, which occurs in primary lung cancers but not in normal cells, activates c-Abl (Fig. 1). FUS1 expression is lost in a majority of small cell lung cancer (80%) and NSCLC samples (100%), indicating that c-Abl activation is likely a frequent event in these tumors. 93 Consistent with the findings by Lin et al., c-Abl/Arg were activated (assessed by phosphorylation of Crk/CrkL) in A549 NSCLC cells. 94 It is unclear whether loss of FUS1 expression is sufficient to activate c-Abl in lung cancer cells or whether other events, such as phosphorylation, also contribute to activation. Furthermore, it is not known whether Arg also is activated by loss of FUS1 expression. 95
Positive Roles for c-Abl and Arg in Solid Tumor Progression
Breast Cancer
Several distinct breast cancer subtypes have been identified using molecular profiling.96,97 Luminal breast cancers express estrogen and progesterone receptors (ER+, PR+) and thus are treated with selective estrogen receptor modulators (SERMs) such as tamoxifen. ErbB2/Her-2+ breast cancer is treated with the monoclonal antibody, trastuzumab/herceptin, which blocks Her-2 function. Triple-negative breast cancers (basal-like), the most aggressive of the breast cancers, which do not express any of the above markers, are treated with conventional chemotherapeutic agents, and this group of patients has the worst prognosis.96,97
Triple-Negative (Basal-Like) Subtype (ER−, PR−, Her-2−)
Using pharmacological (imatinib) and RNAi approaches, we demonstrated that activation of c-Abl and/or Arg promoted breast cancer cell proliferation, anchorage-independent growth, survival in response to nutrient deprivation, and Matrigel transwell invasion, which has invasive and migratory components.84,98 A pro-proliferative, pro-transforming role for c-Abl/Arg was confirmed by Sirvent and colleagues, 86 who demonstrated that c-Abl/Arg promote anchorage-independent growth and proliferation of BT-549 breast cancer cells via activation of Rac- and Erk5-dependent pathways (Fig. 1). In addition, Smith-Pearson and colleagues 99 showed that activation of c-Abl/Arg was required for Matrigel transwell invasion of MDA-MB-231 cells and highly metastatic variants, matrix degradation, invadopodia formation, and surface MT1-MMP expression (Fig. 1). Moreover, Mader et al. 100 demonstrated that Arg localized to invadopodia and promoted EGFR/Src/cortactin-mediated matrix degradation, actin polymerization, invadopodia formation, and Matrigel transwell invasion of MDA-MB-231 cells, and Sossey-Alaoui and colleagues 101 showed that c-Abl not only promotes matrix degradation but also stimulates PDGF-directed motility of MDA-MB-231 cells via c-Abl–dependent phosphorylation of WAVE3 (Fig. 1). Furthermore, activation of c-Abl by the proangiogenic factor, bFGF, promoted endothelial cell proliferation, survival in response to serum-deprivation, motility, tube formation, angiogenesis, and tumor growth of a MDA-MB-231 xenograft model. 36 Consistent with the above findings, SKI-606 (bosutinib), a dual SFK/Abl/Arg inhibitor, prevented tumorigenesis, angiogenesis, and metastasis of MDA-MB-231 xenografts; however, the authors did not test whether these effects were c-Abl/Arg dependent (downstream of SFKs and/or by direct c-Abl/Arg inhibition). 102 Thus, there are numerous examples demonstrating that active c-Abl/Arg promote invasion and matrix degradation of triple-negative breast cancer cells and thus are likely to promote metastatic progression. However, there are no animal studies to date that directly test this hypothesis, and to date, there are no data demonstrating a correlation between c-Abl/Arg activity and grade and/or stage of primary, triple-negative breast cancers.
ER+/PR+ (Luminal) and Her-2+ Subtypes
There is accumulating evidence that c-Abl and Arg also promote the growth and progression of anchorage-independent, tumorigenic, ER+/PR+ breast cancer cells. Imatinib inhibited proliferation and soft-agar growth of MCF-7 cells, 86 and c-Abl/Arg promoted MCF-7 xenograft growth (assessed by silencing c-Abl/Arg) by phosphorylating and blocking lysosomal degradation of the antiapoptotic oncogenic protein, galectin-3 (Fig. 1). 103 The data described above were performed in media containing estrogenic factors; thus, estrogen-dependent growth of ER+ breast cancer cells was not assessed. Significantly, Oh and colleagues 104 showed that a nuclear receptor coactivator, AIB1, which is required for coactivation of ERα and PR promoters and is critical for estrogen and IGF-1–dependent growth, bound and was phosphorylated by c-Abl. Moreover, c-Abl promoted a c-Abl/AIB1/ERα/CBP-p300 complex, and inhibition or silencing c-Abl abrogated PR-dependent gene expression and estrogen-dependent growth of MCF-7 cells, which contain amplified AIB1 (Fig. 1). 104 These data were corroborated by Zhao and colleagues, 87 who demonstrated that c-Abl bound ERα in T47D (ER+, PR+) cells, which express high levels of ERα and c-Abl, and silencing c-Abl with 2 independent siRNAs reduced ER transcriptional activity to a similar level as tamoxifen. Moreover, transfection of wild-type, kinase-inactive, or constitutively active forms of c-Abl but not Arg into MCF-7 cells that expressed low levels of c-Abl enhanced estrogen-mediated reporter activity, indicating that c-Abl promoted ERα activation via a kinase-independent mechanism. 87 He and colleagues 105 also showed that c-Abl bound and phosphorylated ERα, stabilizing ERα, promoting ERα transcriptional activity, and stimulating estrogen-dependent proliferation and soft-agar growth (Fig. 1). However, in this case, enhancement of ER-mediated transcription required c-Abl kinase activity. 105 In primary breast cancers, c-Abl and ERα expression were not correlative 87 ; however, since c-Abl expression is unlikely to reflect activity, more studies are needed to determine whether active c-Abl and ERα are coexpressed in primary ER+ breast cancers. Since c-Abl binds ERα, it is likely to competitively inhibit SERM binding to ERα. Indeed, c-Abl promoted tamoxifen resistance of T47D and BT-474 cells (ER+, ErbB2/Her-2+) but had no effect on MDA-MB-231 cells, which are resistant due to lack of ER expression. 87 Blocking c-Abl activity also sensitized T47D and BT-474 breast cancer cells to fulvestrant, reducing cell viability and preventing cell cycle progression by promoting fulvestrant-induced degradation of ERα. 106
In summary, there is accumulating evidence that activation of c-Abl and/or Arg promotes breast cancer development and progression. However, clearly more studies are needed to determine whether c-Abl and Arg activation occurs in primary breast cancers and correlates with breast cancer progression, and additional animal studies are needed to definitively substantiate a role for c-Abl and Arg in promoting breast cancer development and progression.
Melanoma
A diagnosis of metastatic melanoma is a death sentence, as there has been little improvement in 5-year survival rates over the past 40 years. Although newer biological agents (e.g., vemurafenib, ipilimumab) show promise, they only extend survival by 3 to 4 months, and thus, additional novel therapies are needed.107-110 Using imatinib, nilotinib, and 2 independent siRNAs, we showed that active c-Abl and/or Arg were required for proliferation, survival during nutrient deprivation, and Matrigel invasion of human melanoma cell lines. 83 Significantly, although both c-Abl and Arg were required for invasion, they promoted invasion via different mechanisms. Whereas c-Abl promoted invasion by increasing MMP-1 transcription via STAT3, Arg promoted invasion by increasing MMP-1, -3, and MT1-MMP transcription independent of STAT3 (Fig. 1). 83 Significantly, inhibition of c-Abl/Arg with nilotinib, but not imatinib, dramatically inhibited melanoma lung colonization in a xenograft experimental metastasis model (assessed by IVIS imaging over time), indicating that nilotinib is a less toxic, more active agent for inhibiting active c-Abl/Arg in vivo. 83 Furthermore, IVIS metastatic fluorescence correlated with c-Abl/Arg activity (pCrk/CrkL staining) in lung lesions from treated animals, indicating that nilotinib’s anti-metastatic capability is linked to c-Abl/Arg inhibition. 83 Consistent with our data, c-Abl/Arg were shown to be activated in metastatic murine melanoma cells (B16F10) relative to its nonmetastatic counterpart (B16F0), and silencing c-Abl/Arg inhibited invasion and matrix degradation. 99 Moreover, imatinib inhibited B16F10 tumor growth, 111 imatinib cooperated with dacarbazine to inhibit B16F10 metastatic progression, 112 and dasatinib (SFK/Abl/Arg inhibitor) inhibited invasion and migration of human melanoma cell lines. 113 However, the dependence of these effects on c-Abl/Arg has not yet been evaluated.
Lung Cancer
Lung cancer is the leading cause of cancer deaths in the United States. Non–small cell lung cancer constitutes 85% of lung cancers, whereas small-cell disease represents the other 15%. Non–small cell lung cancer is often diagnosed at a late stage and thus has a poor prognosis. 114 Activation of c-Abl, mediated by loss of FUS1 expression, promoted anchorage-independent growth of non–small cell lung cancer cells, and FUS1 reexpression inhibited c-Abl expression and kinase activity and dramatically induced apoptosis (Fig. 1). 92 In addition, amplification of the c-Abl/Arg substrate, CrkL, promoted proliferation, survival, motility, and invasion of lung cancer cells. 115 Furthermore, Crk expression and phosphorylation were increased in poorly differentiated as opposed to well-differentiated lung adenocarcinomas. 116 Since phosphorylation of Crk on Y251 promotes c-Abl activation, 20 increased Crk/CrkL expression also might contribute to c-Abl/Arg activation in lung cancer cells and promote progression; however, this remains to be tested. In addition to promoting anchorage-independent growth, activation of c-Abl/Arg in A549 lung cancer cells also promotes autophagy and, thus, degradation of long-lived proteins, which likely allows lung cancer cells to survive nutrient deprivation. 94
Gastric and Liver Cancers
Although gastric cancer diagnoses have decreased in the United States due to effective treatment of Helicobacter pylori, it remains the second most common cause of cancer-related deaths worldwide, and overall survival of metastatic gastric cancer is dismal.117,118 Liver cancer often occurs following chronic liver disease, and treatment is limited to surgical resection, liver transplant, radiofrequency ablation, and transarterial chemoembolization. 119 Sustained activation of c-Abl, downstream of constitutively active c-Met, promoted survival during nutrient deprivation, anchorage-independent growth, and tumorigenesis in gastric and/or liver cancer cells. 89 Interestingly, activated c-Abl induced p38 phosphorylation and subsequent phosphorylation of p53 (S393), leading to increased p53 transcriptional activity and upregulation of p53 targets (Mdm2) (Fig. 1). 89 Moreover, phospho-c-Met expression correlated with phospho-p53 (S392) and Mdm2 levels in primary hepatocellular carcinomas; however, an association between c-Abl/Arg activation and c-Met, p53, and Mdm2 was not tested. 89 c-Abl activation in liver cancer cells also was shown to promote progression, as activation of c-Abl downstream of claudin-1 increased migration and Matrigel invasion via a PKCδ/MMP-2–dependent pathway (Fig. 1). 90
Prostate Cancer
Despite advances in prostate cancer detection (prostate-specific antigen [PSA]) and Gleason grading, prostate cancer is still the second most common cause of cancer deaths in men as there are few drugs to treat metastatic, castration-resistant disease. 120 Inhibition of SFKs and c-Abl/Arg with bosutinib blocked migration, invasion, anchorage-independent growth, and proliferation of PC3 and DU-145 prostate cancer cells, which was accompanied by decreased Akt, ERK1/2, and FAK phosphorylation. 121 Furthermore, bosutinib inhibited PC-3 tumor growth initiated by subcutaneous or intraskeletal injection. 121 Silencing Src produced similar effects as bosutinib; however, the authors did not investigate whether the effects also could be mediated by Src-dependent activation of c-Abl/Arg and/or due to direct inhibition of c-Abl/Arg by bosutinib. Bone metastases, which are common for prostate and breast cancers, increase morbidity and mortality. Significantly, dasatinib treatment or silencing c-Abl or Src inhibited osteoblast proliferation and promoted osteoblast differentiation, indicating that c-Abl inhibition may prevent bone metastatic growth. 122 c-Abl–dependent phosphorylation of WAVE3, which is upregulated in advanced tumors and promotes metastasis, increased prostate cancer cell invasion, indicating a role for c-Abl in prostate cancer progression. 123 Moreover, activation of c-Abl by PDGF promoted prostate cancer cell survival by inducing expression of the antiapoptotic protein, MCL-1, via a p68/β-catenin signaling pathway (Fig. 1). 124 c-Abl/Arg also play a role in motility/invasion of prostate cancer cells, as c-Abl/Arg–mediated phosphorylation of the carbohydrate-binding protein, galectin-3, prevented galectin-3 cleavage by PSA, increasing full-length extracellular galectin-3, which promoted wound healing and Matrigel invasion.125,126 Thus, there is accumulating evidence that activation of c-Abl/Arg promotes prostate cancer progression.
Thyroid, Colon, Rhabdoid, and Ovarian Cancers
In anaplastic thyroid cancer cell lines, activation of c-Abl increased cell cycle progression and tumorigenesis. 91 In colon cancer cells, c-Abl promoted epithelial-mesenchymal transition (EMT) downstream of PDGF by phosphorylating p68RNA helicase in the nucleus, thereby inducing β-catenin activation (Fig. 1). 127 Using pharmacological and RNAi approaches, activation of c-Abl in atypical teratoid/rhabdoid tumors was shown to increase cell proliferation. 82 Furthermore, dasatinib (SFK/Abl/Arg inhibitor) induced cell death and prevented tumorigenesis of ovarian cancer cells; however, the dasatinib target(s) was not identified. 128 In contrast, Le and colleagues 129 definitively showed that c-Abl and Src are activated in ovarian cancer cell lines, and dasatinib treatment or silencing c-Abl sensitized ovarian cancer cells to paclitaxel, inhibiting colony formation and xenograft growth.
Does c-Abl Prevent Solid Tumor Progression in Some Cell Contexts?
Although the vast majority of evidence indicates that c-Abl/Arg activation promotes solid tumor development and/or progression, some data suggest that c-Abl may suppress tumorigenesis or progression in some cell contexts. For example, treatment of MDA-MB-435 cells with ephrin B2, which transiently activates EphB4 and c-Abl/Arg, inhibits cell proliferation, 3D growth, invasion, MMP-2 expression, and xenograft tumor growth, and these effects are blocked by imatinib. 130 Thus, the authors suggest that c-Abl/Arg mediate the tumor-suppressive effects of ephrinB2/EphB4; however, the dependence on c-Abl/Arg was not confirmed using other approaches (e.g., RNAi), and the effects on progression (metastasis) were not investigated. 130 Furthermore, the physiological relevance of these data is unclear since most cancer cells do not secrete ephrin B2. 130 In contrast, using a highly metastatic derivative of the same cell line (MDA-MB-435s/M14), imatinib, nilotinib, and 2 independent siRNAs, we found that c-Abl and/or Arg promote proliferation in serum and serum-free conditions, survival following nutrient deprivation, and Matrigel transwell invasion toward IGF-1.83,84,98 Furthermore, inhibition of c-Abl/Arg activity with nilotinib prevented experimental metastasis of MDA-MB-435s xenografts. 83 Similarly, overexpression of Rin1, a tumor suppressor whose expression is lost in cancer cells, suppresses MDA-MB-231 invasion by binding and activating Arg. 131 These results contrast with 2 reports demonstrating that in the absence of Rin1 overexpression, Arg promotes invasion and matrix degradation of MDA-MB-231 cells (using RNAi and pharmacological approaches).99,100 Taken together, these data suggest that ephrin B2 or Rin1 expression may suppress tumorigenicity in an c-Abl/Arg–dependent manner in nontransformed cells that express ephrin B2 and/or Rin1; however, during cancer progression, ephrin B2 and Rin1 expression are lost, c-Abl/Arg are activated by other signals (e.g., growth factors/SFKs, loss of inhibitor expression), and sustained active c-Abl/Arg are potent promoters of tumorigenesis and/or metastasis. Clearly, additional experiments are needed to directly test this hypothesis.
TGF-β inhibits cancer development and thus is considered a tumor suppressor; however, during cancer progression, TGF-β promotes EMT, which is characterized by loss of epithelial markers, gain of a mesenchymal phenotype, and increased invasive potential. 132 To test whether c-Abl regulates breast cancer EMT, Allington and colleagues 133 expressed an activated form of c-Abl and silenced or inhibited c-Abl in murine nontumorigenic, NuMG epithelial cells and in tumorigenic murine 4T1 mesenchymal-like cells. Interestingly, treatment of NMuMG cells, which likely express basal, regulated c-Abl (assessed by ATP consumption assay), with high concentrations of imatinib (17-50 µM) or silencing c-Abl promoted morphological EMT. 133 Conversely, transfection of a constitutively active form of c-Abl into tumorigenic 4T1 cells promoted a epithelial-like morphology and inhibited tumor growth, whereas low concentrations of imatinib had no effect on 4T1 tumor growth. 133 Moreover, expression of a kinase-inactive form of c-Abl (which might not act as a dominant-negative since c-Abl has kinase-independent functions)134-136 promoted invasion toward TGF-β, whereas expression of a constitutively active form prevented TGF-β–induced invasion, MMP expression, and proliferation. 133 These results contrast with data in human cancer cells demonstrating that inhibition/silencing of c-Abl and/or Arg inhibits matrix degradation, MMP activation, invasion, and PDGF-induced EMT.84,86,90,98-100,103,127 In addition, Wilkes and Leof 37 demonstrated that TGF-β induced c-Abl activation in murine and human mesenchymal cells and promoted anchorage-independent growth in an Abl-dependent manner. The conflicting results obtained may be due to differences in cell lines used, since responses to TGF-β vary dramatically between cell culture models. 37 Alternatively, the opposing results also could be due to differences in the activation status of c-Abl. c-Abl/Arg are highly activated in the human cancer cells used84,86,98-100,103; however, in vitro kinase assays and/or Crk/CrkL phosphorylation were not used to determine the activation status of c-Abl in NuMG or 4T1 cells, although ATP consumption assays suggested that c-Abl/Arg activity is likely low in NuMG cells. 133 Thus, regulated, endogenous c-Abl may serve to restrict tumorigenesis, whereas sustained active c-Abl may promote tumor progression. Opposing roles for regulated and activated c-Abl also have been observed in response to DNA damage (below) and during cell-cell adhesion. 72 Interestingly, expression of constitutively active c-Abl in 4T1 cells dramatically reduced endogenous c-Abl expression; thus, the effects observed with this mutant could also be due to loss of endogenous c-Abl. 133 Moreover, an effect on the end point of EMT, metastasis, was not tested. Thus, clearly more studies are needed to validate a role for c-Abl in suppressing EMT and metastatic progression, and the role that Arg plays in this process also needs to be evaluated.
Conflicting data also have been obtained in thyroid cancer cells. Imatinib promoted HGF-induced motility of papillary thyroid cancer cells. 137 In contrast, Podtcheko and colleagues 91 showed that c-Abl was activated in anaplastic but not in papillary thyroid carcinoma cells, and imatinib inhibited anaplastic cell cycle progression and tumorigenesis. Unfortunately, neither study used RNAi nor rescue approaches to definitively identify the imatinib target. However, since c-Abl is activated in anaplastic but not papillary thyroid cancer cells, it is plausible that active c-Abl might drive anaplastic tumorigenesis, while basal, endogenous c-Abl may serve to restrict tumor growth in papillary cancers where c-Abl is not activated. Imatinib also promoted HGF-induced cell scattering, foci formation, and cell motility of HeLa cells. 62 This contrasts with data in gastric and liver cancer cells, demonstrating that sustained activation of c-Abl by a constitutively active HGF receptor (c-Met) promoted anchorage-independent growth and tumorigenesis. 89 Again, it is plausible that sustained activation of c-Abl promotes tumor progression in gastric/liver cancer cells, whereas basal, regulated c-Abl (e.g., HeLa cells) has opposing effects. Alternatively, c-Abl might restrict migration when cells need to proliferate (at the ectopic site) and block proliferation when motility is required (during intravasation/extravasation). Thus, the cellular milieu/tumor microenvironment is likely to be extremely important in determining whether c-Abl functions to promote or suppress tumor growth and/or progression. Importantly, if c-Abl indeed acts as a tumor or metastasis suppressor in some cancer types, one would expect to observe decreased expression and/or activation of c-Abl in primary tumors relative to normal tissue, and to date, this has not yet been documented. Clearly, additional studies are needed to solidify a role for c-Abl in tumor suppression.
Numerous proteins other than c-Abl also have been shown to act in a “bipolar” fashion. For example, Rac1 promotes epithelial cell-cell adhesion, which restricts cell movement; however, during EMT, activated Rac promotes dissolution of adherens junctions and increases cell motility. 138 Likewise, the β1 integrin is required for tumor progression but also inhibits cancer progression. 139 Since manipulating c-Abl/Arg expression and activity potently affects tumorigenesis and progression, these proteins clearly are critical regulators. Understanding the signals that switch c-Abl from a suppressor of tumorigenesis to a promoter of solid tumor progression (such as by loss of Rin1 and ephrin B2 expression) will be critical goals for future research. Importantly, since inhibition of endogenous, inactive c-Abl may promote tumor progression, clinical studies using c-Abl/Arg inhibitors must be restricted to tumors containing highly active c-Abl/Arg (see below).
c-Abl/Arg and Drug Resistance
In nontransformed cells, treatment with chemotherapeutic DNA-damaging agents activates the nuclear pool of c-Abl, which promotes cell cycle arrest and/or apoptosis. 51 Constitutively active Abl proteins (e.g., v-Abl, BCR-Abl), which promote proliferation, survival, and motility, are located exclusively in the cytoplasm, and forced expression of BCR-Abl in the nucleus induces apoptosis. 140 In metastatic MDA-MB-435s/M14 and MDA-MB-231 cancer cells, active c-Abl is primarily cytoplasmic, similar to BCR-Abl and v-Abl.85,98 Interestingly, unlike in nontransformed cells, inhibition of active c-Abl with imatinib sensitizes these cells to a variety of chemotherapeutic agents, indicating that active cytoplasmic c-Abl/Arg likely induces resistance rather than apoptosis in response to DNA-damaging agents. 85 Imatinib also sensitizes CML, head and neck, ovarian, colon, non–small cell and small cell lung cancer, glioma, and neuroblastoma cell lines to chemotherapeutic agents141-150; however, the targets of imatinib were not identified in these reports. In contrast, Le and colleagues 129 screened an siRNA kinase library to identify kinases whose decreased expression increased paclitaxel-mediated antiproliferative effects in ovarian cancer cells and identified SFKs (Src, Fyn, Yes) as well as c-Abl and Arg. The authors validated their findings using siRNAs and also demonstrated that silencing or inhibiting Src or c-Abl sensitized ovarian cancer cells to paclitaxel, inhibiting colony formation, inducing apoptosis and mitotic catastrophe, and preventing xenograft tumor growth. 129 Thus, transient activation of nuclear, regulated c-Abl by DNA-damaging agents in nontransformed cells induces apoptosis, whereas sustained activation of cytoplasmic c-Abl/Arg in solid tumor cells promotes resistance to the same agents. Significantly, Yoshida and colleagues 45 presented data that may explain this dichotomy. In nontransformed cells, c-Abl binds 14-3-3 proteins, and following DNA damage, 14-3-3 is phosphorylated, which releases c-Abl, targeting it to the nucleus. In cancer cells, the oncoprotein, MUC1, is overexpressed, and c-Abl binds and phosphorylates MUC1, which inhibits the interaction between c-Abl and 14-3-3 proteins. Therefore, MUC1 permanently sequesters c-Abl in the cytoplasm, preventing targeting of c-Abl into the nucleus following DNA damage. 46 In summary, since inhibition of c-Abl/Arg sensitizes solid tumor cells to conventional agents, utilization of Abl inhibitor/chemotherapeutic drug combinations may be more effective than either drug alone for the treatment of solid tumors expressing activated c-Abl/Arg (see below).
Clinical Targeting of Abl Family Kinases in Solid Tumors
Numerous clinical studies have been initiated using c-Abl/Arg inhibitors such as imatinib, nilotinib, dasatinib, and bosutinib (SKI-606) for the treatment of solid tumors.151-179 Some trials targeting tumors with activation of c-Kit or PDGFR (other imatinib, nilotinib targets) demonstrated effectiveness152,180,181; however, the majority of trials were not targeted. Despite lack of targeting, clinical activity was noted for imatinib in anaplastic thyroid cancer 171 ; dasatinib + dacarbazine had activity in two-thirds of metastatic melanoma patients lacking c-Kit mutations 174 ; dasatinib showed activity in advanced Her-2+ breast cancer, 175 metastatic breast cancer, 178 non–small cell lung cancer,182,177 and metastatic prostate cancer 179 ; and produced partial responses and/or stabilized disease in a small subgroup of patients with triple-negative breast cancer. 183 Bosutinib also had activity in metastatic breast cancer, particularly in ER+/PR+ patients. 184 However, many nontargeted trials did not demonstrate effectiveness, and thus, it has been suggested that these trials failed because c-Abl and/or Arg inhibit rather than promote tumorigenesis. 185 However, since inhibition of inactive c-Abl may have opposite effects from inhibiting sustained, active c-Abl/Arg, and c-Abl/Arg are not likely to be activated in all tumors of a given type, it is not possible to accurately evaluate the efficacy of c-Abl/Arg inhibitors in nontargeted trials. The importance of using a targeted population when testing signaling inhibitors cannot be understated. Sledge and Miller 186 stated this point elegantly: “A relatively inactive agent in an unselected population of breast cancer patients may be a highly active agent in a tightly defined subpopulation (if we just knew the proper definition). One only has to look at the history of trastuzumab—active in Her-2-positive patients, inactive in Her-2 negative—to realize that we may have tossed away numerous potentially useful agents over the years through inadequate targeting.” In addition, our data indicate that effective inhibition of sustained, active c-Abl/Arg requires higher doses of imatinib than inhibition of BCR-Abl in vitro, and higher doses were toxic in vivo. 83 Thus, imatinib may not be the optimal drug for inhibiting c-Abl/Arg in tumors expressing nonmutated, active forms of c-Abl/Arg, and second-generation drugs that are more sensitive (e.g., nilotinib) are likely to be more effective. 83 Furthermore, many trials using c-Abl/Arg inhibitors were conducted on patients who failed prior treatment regimens, and thus, these tumors are likely to have upregulated ABC transporters. Since ABC transporters are known to efflux c-Abl/Arg inhibitors, this also could explain trial ineffectiveness.187-189 Interestingly, SFK/Abl/Arg inhibitors appear to have had more success in nontargeted trials compared with imatinib. This may be because these agents inhibit upstream activators of c-Abl/Arg (SFKs) in addition to inhibiting c-Abl/Arg and thus more effectively target c-Abl/Arg–dependent pathways. Alternatively, these drugs may inhibit tumors containing activated SFKs, activated c-Abl/Arg, or activated SFK AND Abl kinases, and thus, the percentage of tumors with activated targets may be higher than for trials using c-Abl/Arg inhibitors, thus making it more likely to observe benefit in an nontargeted trial. Additional experiments aimed at evaluating the activation status of SFKs and c-Abl/Arg prior to and following treatment with these agents clearly are needed to test these possibilities.
To determine whether c-Abl/Arg inhibition is effective for treating solid tumors, there must be an effective, reliable way to ascertain c-Abl/Arg activity in solid tumors. One possibility may involve staining tumor serial sections with antibodies directed against (1) c-Abl and Arg to examine overall expression, (2) c-Abl/Arg substrates (e.g., Crk/CrkL, cortactin, WAVE3, galectin-3)83,100,101,103,123,125,126 to indirectly examine kinase activity, and (3) proteins known to act downstream of c-Abl/Arg in a particular cancer type (e.g., STAT3, p38, p68, ERK5).83,86,89,127 Alternatively, for tumor types containing low levels of stromal tissue and other non-tumor-associated cells, direct assessment of activity could be determined by kinase assay on tumor homogenates if enough sample is available.190,191 Clearly, the ability to effectively identify patients who may benefit from c-Abl/Arg inhibitor treatment is critical for determining whether these drugs have clinical efficacy.
Summary
Accumulating evidence suggests that c-Abl and Arg not only are important for the development and progression of leukemias but also are involved in the progression of solid tumors. Future research will need to be focused on developing new ways of identifying tumors containing active c-Abl and/or Arg. It also will be important to identify downstream signaling pathways and determine whether these are tissue specific or are conserved across multiple cancer types. It also is critical to determine whether the roles of c-Abl and Arg are distinct, overlapping, or antagonistic since drugs targeting these molecules inhibit both kinases. Furthermore, future experiments aimed at identifying the signals that convert c-Abl from a tumor suppressor to a tumor promoter are needed so that cancers that are likely to be inhibited by anti-Abl/Arg therapies can be identified. Finally, additional animal studies, preferably using spontaneous models, clearly are warranted to determine the extent to which c-Abl/Arg are involved in solid tumor development and progression.
Footnotes
Acknowledgements
We thank Jonathan Sims for useful discussions during the writing of this manuscript.
Declaration of Conflicting Interests
Dr. Plattner’s work is funded by the National Institutes of Health. The authors declare no other potential conflict of interest with respect to the authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported, in part, by a grant from the National Cancer Institute 1R01CA116784.