
Abstract
Regulation of Myc protein abundance is critical for normal cell growth as evidenced by the fact that deregulated Myc expression is a hallmark of many cancers. One of several important mechanisms that control Myc levels involves its phosphorylation-dependent proteolysis. Previous studies have shown that phosphorylation of threonine 58 by glycogen synthase kinase 3β (GSK3β) within the conserved Myc Box I sequence results in binding by the ubiquitin ligase Fbw7-SCF complex, followed by ubiquitination and proteasome-mediated degradation of Myc. Here, we show that induction of Myc in several cell types correlates with loss of the inhibitory serine 9 phosphorylation of GSK3β and its increased kinase activity. The Myc-induced decrease in serine 9 phosphorylation is blocked by okadaic acid, an inhibitor of protein phosphatase 2A (PP2A). We therefore examined components of PP2A complexes and found that, among the regulatory B56 subunits, only the promoter of the ppp2r5d gene, encoding the B56δ isoform, is directly bound and transcriptionally activated by Myc in an E-box–dependent manner. Furthermore, we find that B56δ associates with both GSK3β and Myc, resulting in phosphorylation of Myc threonine 58, the well-established signal for ubiquitination and degradation. Furthermore, overexpression, or siRNA-mediated knockdown, of B56δ respectively results in accelerated, or retarded, rates of Myc degradation. Together, our data indicate that Myc limits its own abundance through a negative feedback pathway involving PP2A and GSK3β.
Introduction
The c-Myc protein, a member of the basic helix-loop-helix zipper (bHLHZ) class of transcription factors, has been shown to directly regulate the expression of several thousand genes involved predominantly in cell growth, proliferation, and metabolism.1,2 In addition, Myc has been reported to possess nontranscriptional functions, having been linked to DNA replication 3 and, as the calpain cleavage product Myc-nick, to cytoplasmic tubulin acetylation and differentiation. 4 Given the large number of targets regulated by Myc, it is not surprising that Myc protein abundance is itself under tight control.5,6 Indeed, Myc abundance has been shown to be regulated at multiple levels, including myc gene transcription, RNA stability, translation, and protein stability. The critical importance of this multilevel regulation is underscored by the fact that Myc is frequently overexpressed and deregulated in a wide range of human and other animal cancers.6,7
The control of Myc abundance through protein degradation has attracted considerable interest. Multiple ubiquitin ligases have been demonstrated to recognize distinct regions of Myc, leading to ubiquitylation and subsequent degradation by the proteasome.8-14 Moreover, Myc proteins in some cancers have been shown to possess increased stability.15-18 Particular interest has focused on the Myc Box I (MBI) region of c-Myc, a highly conserved N-terminal segment containing the sequence P(57)-T-P-P-L-S-P(63) that comprises a phosphodegron. Phosphorylation of serine 62 (S62) leads to increased Myc stabilization but also acts as to prime phosphorylation, permitting glycogen synthase kinase 3β (GSK3β) to phosphorylate threonine 58 (T58), which in turn leads to Myc degradation.17,19-21 The GSK3β-mediated phosphorylation of c-Myc at T58 in MBI provides a direct binding site for the F-box protein Fbw7, resulting in ubiquitination followed by proteasomal degradation of c-Myc.11,12,22 These phosphorylation events appear to link Myc stability to cellular signal transduction pathways. For example, S62 phosphorylation is mediated by Ras/ MAPK and Cdk1.20,21,23 Moreover, GSK3β is regulated through multiple pathways.24,25 Several of these, including phosphoinositide 3-kinase (PI3K)–AKT (PKB), p90RSK, p70S6K, and PKA lead to inhibitory phosphorylation at GSK3β serine 9 (S9).25-27
The studies described above indicate that the interplay between phosphorylation events at S62 and T58 within c-Myc MBI is involved in the regulation of c-Myc abundance. Recent work has shown that the phospho-T58: phospho-S62 ratio is controlled by a protein complex involving the scaffolding protein axin that appears to coordinate binding of GSK3β, Pin1 prolyl isomerase, and protein phosphatase 2A (PP2A)–B56α.18,28 PP2A is an abundant protein phosphatase existing as heterotrimeric complexes of C (catalytic) and A subunits and one of a large number of B subunits that act to regulate and target the complex. 29 PP2A-B56α has been shown to dephosphorylate c-Myc S62, resulting in decreased stability and altered transcriptional activity of the c-Myc protein.30,31 It has been reported that the tumor suppressor Axin1 facilitates formation of a GSK3β-Pin1–B56α complex associated with Myc degradation 28 and that loss of complex formation is associated with increased Myc stability and oncogenicity in breast cancers. 18
The connections between Myc protein stability and GSK3β activity prompted us to ask whether Myc itself, as part of its regulatory circuitry, might control the activity of GSK3β. Here, we report that c-Myc is linked to GSK3β activation through induction of B56δ, a PP2A regulatory subunit and a transcriptional target of c-Myc.
Results
c-Myc induction results in activation of GSK3β
Because GSK3β directly phosphorylates c-Myc, leading to Fbw7-SCF binding and c-Myc protein degradation, we asked whether there exists a feedback pathway through which c-Myc influences GSK3β abundance or activity. We began by employing the P493-6 human B cell line with a conditional c-myc allele, whose expression is repressed in the presence of tetracycline (Tet) and activated in the absence of Tet. Growth and proliferation of P493-6 cells have been shown to be strictly dependent on the induction of c-myc.32,33 Phosphorylation of S9 in GSK3β is a well-established inhibitory phosphorylation that converts the N-terminus of the enzyme into a pseudosubstrate, thereby blocking normal access of substrates to the catalytic core. 34 Using a specific antibody that recognizes this modification, we find a striking increase in GSK3β phospho-S9 when the c-Myc protein is downregulated following the addition of increasing amounts of Tet to the culture medium (Fig. 1A). Conversely, P493-6 cells maintained in the presence of Tet show, upon removal of Tet, robust induction of c-Myc and dephosphorylation of GSK3β S9 (Suppl. Fig. S1A). In both cases, no change is detected in total GSK3β protein levels (Fig. 1A and Suppl. Fig. S1A). To determine whether the diminished S9 phosphorylation occurred as a result of the induction of cell proliferation, we removed serum for 24 hours and 48 hours from P493-6 cells in the presence or absence of Tet. Previous studies have demonstrated that cell proliferation subsequent to the induction of c-Myc requires serum. 33 Figure 1B shows that GSK3β S9 phosphorylation is decreased upon c-Myc induction whether or not serum was present in the culture medium, although the decrease is somewhat attenuated in the absence of serum. This suggests that c-Myc–mediated proliferation may contribute to, but is not absolutely required for, loss of GSK3β inhibitory phosphorylation. We also found that the anticorrelation between c-Myc and GSK3β S9 phosphorylation was not restricted to B cells. When we performed an siRNA knockdown of c-Myc in the 293T epithelial cell line, we again observed increased GSK3β S9 phosphorylation upon downregulation of c-Myc (Fig. 1C).
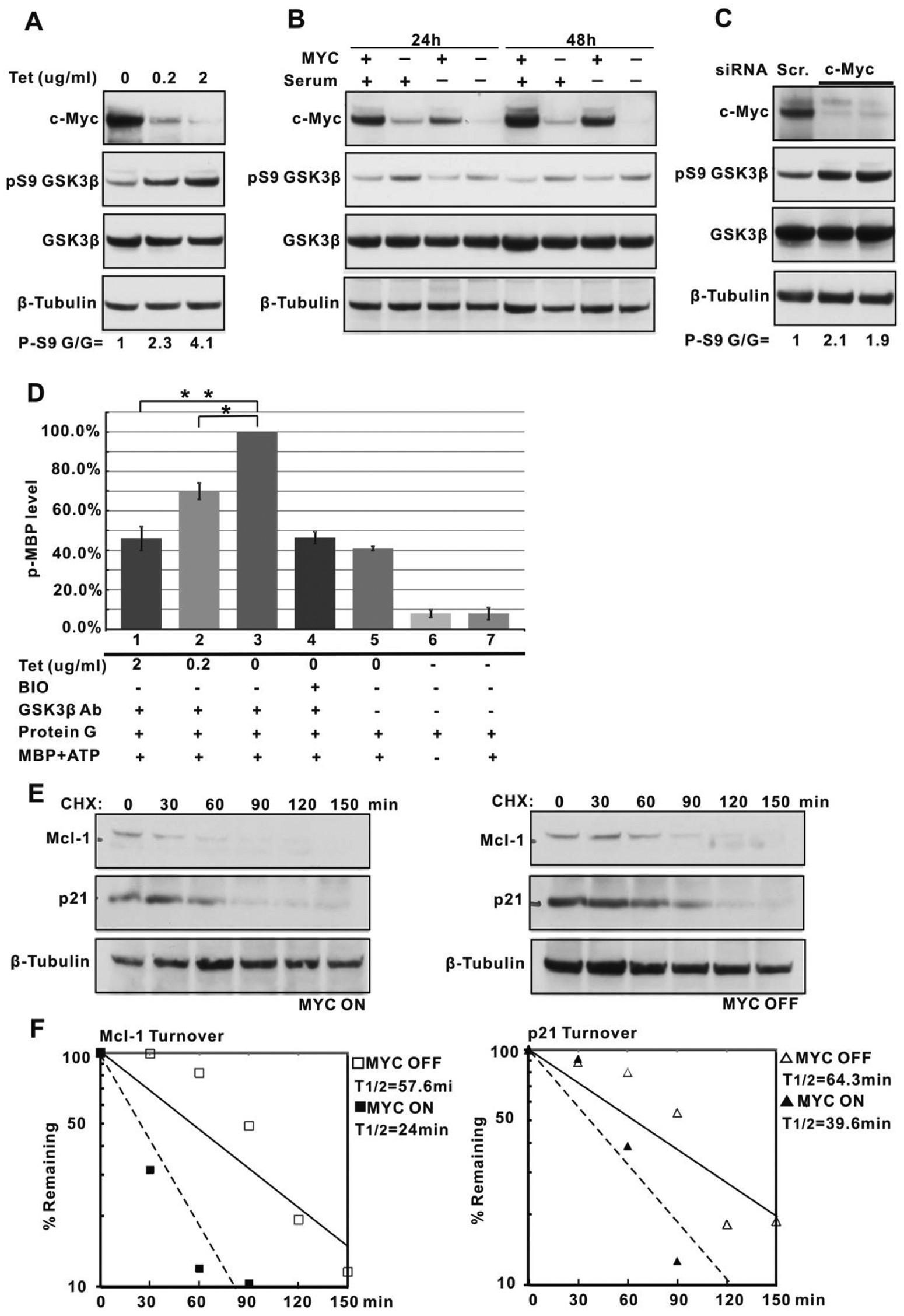
GSK3β activity following induction of c-Myc expression. (
To directly assess whether loss of S9 phosphorylation results in increased GSK3β kinase activity under our conditions of c-Myc induction, we immunopurified GSK3β from P493-6 cells and performed an in vitro kinase assay using an unphosphorylated myelin basic protein (MBP), a standard GSK3β substrate. 35 As shown in Figure 1D, GSK3β derived from P493-6 cells grown in decreasing amounts of Tet displayed significantly increased kinase activity. This activity was inhibited by treatment with the specific GSK3β inhibitor BIO. 36 Moreover, nonspecific IgG, or controls lacking antibody, displayed low or no activity.
To determine the consequences of GSK3β activation following upregulation of c-Myc in P493-6 cells, we examined the half-lives of the antiapoptotic protein Mcl-1 and the p21CIP1 cyclin-dependent kinase inhibitor. Both of these proteins are known to be phosphorylated by GSK3β to trigger their proteasome-mediated degradation.37,38 We carried out cycloheximide chase experiments in Tet-treated (Myc-off) and untreated (Myc-on) P493-6 cells. As depicted in Figure 1E and 1F, the half-lives of both proteins are significantly reduced following c-Myc induction, a result consistent with the loss of GSK3β inhibitory phosphorylation and increased kinase activity.
PP2A involvement in GSK3β activation following the induction of c-Myc
Inhibitory GSK3β S9 phosphorylation has been shown to be mediated during early G1 phase by Akt through the PI3K pathway.26,39,40 However, upon Tet-induced downregulation of c-Myc, and increased GSK3β S9 phosphorylation, we were unable to detect increased phospho-T308 AKT, which activates phosphorylation for AKT (Suppl. Fig. S1B). Moreover, when we inhibit PI3K with LY294002, we observed decreased AKT phospho-T308 levels as expected but nonetheless detected a decrease in GSK3β S9 phosphorylation when c-Myc is induced (Suppl. Fig. S1B). These data suggest that AKT activity does not fully account for the decreased GSK3β activity observed upon Myc repression. We noted however that treatment with okadaic acid (OA), an inhibitor of PP2A at nanomolar concentrations, 41 resulted in increased levels of GSK3β S9 phosphorylation in both Myc-uninduced and -induced contexts (Fig. 2A and Suppl. Fig. S1B). Indeed, the levels of GSK3β phospho-S9 remained as high in Myc-induced P493-6 cells in the presence of OA as in Myc-uninduced cells (Fig. 2A).
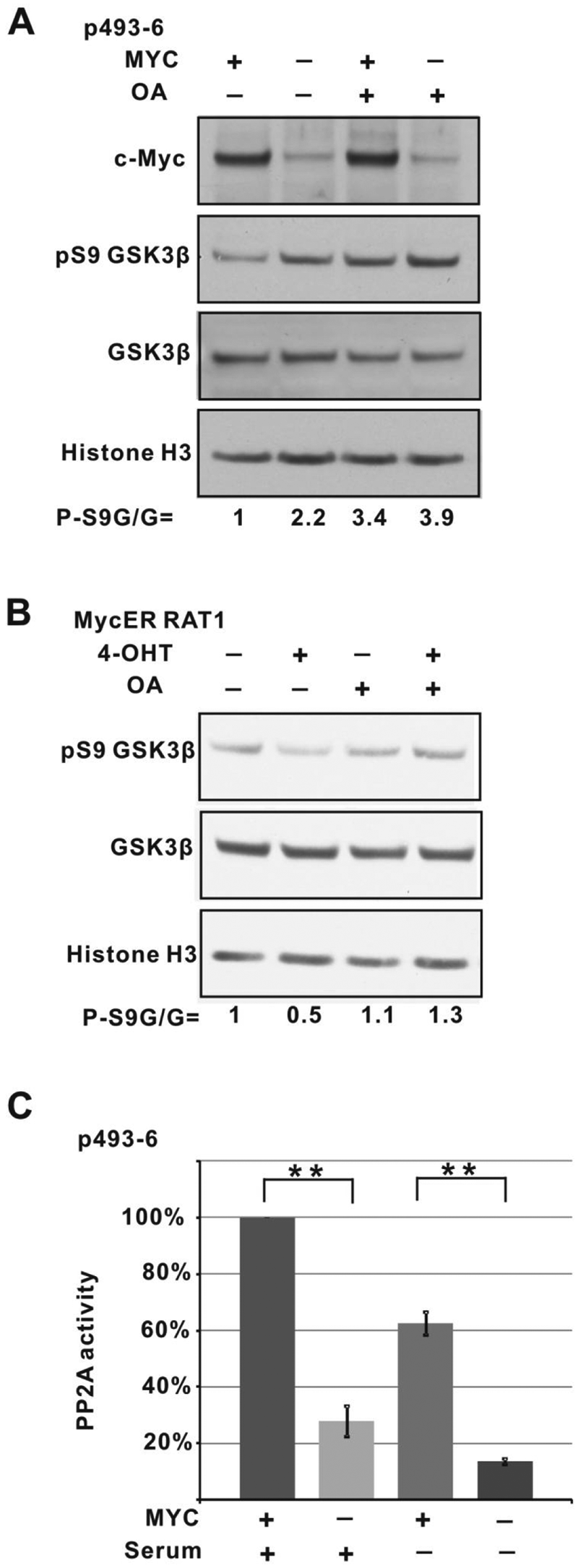
PP2A is involved in c-Myc–mediated GSK3β dephosphorylation. (
We extended this observation by examining the Rat1 fibroblast cell line expressing a Myc-ER fusion protein that is activated and translocated to the nucleus following treatment with 4-OH-tamoxifen (4-OHT) (Suppl. Fig. S2).42,43 Under these conditions, we again observe decreased GSK3β S9 phosphorylation upon 4-OHT treatment. Treatment with 10 nM OA blocks this 4-OHT/Myc–induced decrease (Fig. 2B). The involvement of PP2A in GSK3β activation through S9 dephosphorylation has been well established and is consistent with our findings.44-47
The ability of OA to block GSK3β S9 dephosphorylation upon Myc induction raises the possibility that PP2A is regulated through Myc. To examine this idea more closely, we assayed PP2A activity by spectrophotometric determination of pNPP hydrolysis in cell lysates, derived from Tet-treated or untreated P493-6 cells, in the presence and absence of OA (10 nM). 48 Figure 2C shows a nearly 4-fold decrease in PP2A phosphatase activity in Tet-treated (Myc-off) cells compared to cells with induced Myc. This large difference is at least in part due to proliferation because if the same experiment is carried out in the absence of serum, the Myc-on cells have approximately 60% of the activity as Myc-on cells grown in serum. Nonetheless, upon Myc downregulation in serum-depleted cells, there is an approximately 3-fold decrease in PP2A activity (Fig. 2C). This is consistent with our data described above (Fig. 1B), indicating that serum-induced proliferation contributes to, but is not absolutely required for, decreased GSK3β S9 phosphorylation upon Myc induction.
Myc directly induces transcription of PP2A subunits
Previous studies have shown that PP2A and its B56 regulatory subunits contribute to the activation of GSK3β.44,49 To determine whether the change in PP2A activity following modulation of Myc levels reflects alterations in the abundance of PP2A regulatory B subunits, we prepared immunoblots of P493-6 Myc-on and Myc-off cells using antibodies against the PP2A catalytic C subunit and against common epitopes in the group of B56 regulatory subunit isoforms.50-52 In this experiment, we observed decreased c-Myc protein levels upon Tet treatment but no change in the PP2A C subunit (Fig. 3A). However, while several of the B56 isoforms (apparent molecular weights >50 kDa) also showed no change, the isoform with the highest apparent molecular weight was decreased in the Myc-off cells (Fig. 3A). The largest B56 subunit, B56δ,53,54 is encoded by the ppp2r5d gene, and we asked whether its transcript levels were influenced by Myc. Using RT-PCR, we found that Tet repression of Myc results in a sharp reduction in ppp2r5d transcript levels (Fig. 3B). Applying quantitative RT-PCR to all 5 of the B56 isoforms showed that only B56δ transcription responded strongly to modulation of c-Myc expression (Fig. 3C).
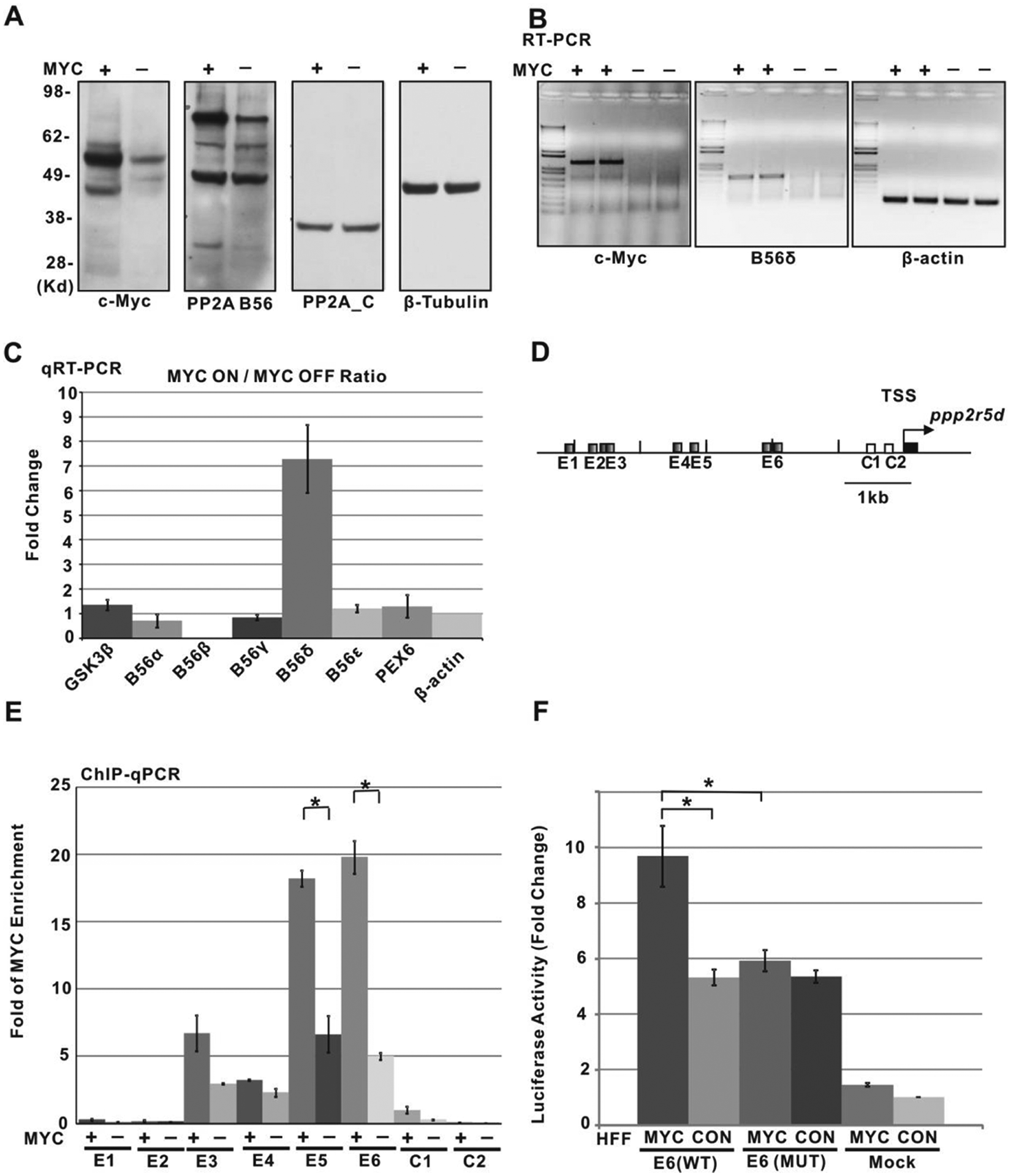
PP2A regulatory subunit B56δ is directly regulated by c-Myc. (
The robust response of B56δ transcription to changes in c-Myc prompted us to ask whether c-Myc might directly bind and regulate ppp2r5d/B56δ gene expression. Because Myc-Max heterodimers regulate transcription through E-box binding sites, 55 we performed quantitative chromatin immunoprecipitation (qChIP-PCR) with anti-Myc to assess Myc association with consensus E-boxes located in the promoter region of ppp2r5d (Fig. 3D and 3E). The results show significant Myc binding at the E5 and E6 noncanonical E-box–containing segments located in the 2- to 3.2-kb region 5′ of the transcription start site (TSS). Weaker (E3, E4) or no (E1, E2) binding is detected at sites further upstream or at non–E-box–containing sites (C1, C2) near the TSS. Importantly, downregulation of Myc in the presence of Tet results in a decreased Myc binding signal from these E-box sites (Fig. 3E). We next asked whether the ppp2r5d promoter region containing the E6 E-box sequences is sufficient to drive Myc-dependent transcription. The region 2.4 kb upstream of the ppp2r5d TSS containing 2 E-boxes (denoted E6 in Fig. 3) was linked to a luciferase reporter and transfected into control or Myc-overexpressing HFFs. As shown in Figure 3F, the activity in control cells of the reporter containing the promoter region increased compared to the activity of the reporter lacking the promoter region, most likely due to binding by transcription factors specific to the 2.4-kb region. However, in the cells containing high levels of Myc, we observed a nearly 2-fold increase in luciferase activity. This increase was abolished by mutation of the 2 E6 E-boxes (Fig. 3F). Therefore, Myc augments transcription driven by the ppp2r5d promoter in an E-box–dependent manner. Taken together, our data demonstrate that Myc directly targets the expression of the B56δ PP2A regulatory subunit to stimulate PP2A phosphatase activity.
Functional associations between GSK3β, c-Myc, and PP2A subunit B56δ
Previous studies had demonstrated the association of both GSK3β and the B56α regulatory subunit of PP2A with Myc.18,28,31 Because our data implicate the PP2A regulatory subunit B56δ in the activation of GSK3β, we asked whether B56δ is associated with GSK3β. We therefore immunoprecipitated endogenous GSK3β, B56δ, or endogenous c-Myc with specific antibodies from P493-6 cell lysates and determined, using immunoblots, which other proteins were associated with the immunoprecipitates (see Materials and Methods). In our initial experiments, we detected weak, but specific, association of GSK3β and c-Myc with B56δ immunoprecipitates from P493-6 cells, suggesting low affinity interaction (Suppl. Fig. S3B). In order to stabilize any associations and increase the signal, we used formaldehyde cross-linking of proteins in cell lysates. Under these conditions, we readily detected the association of endogenous c-Myc and GSK3β with overexpressed His-tagged B56δ and B56α (Suppl. Fig. S3A). Moreover, endogenous B56δ and c-Myc were detected in endogenous GSK3β immunoprecipitates, and endogenous B56δ and GSK3β were detected in immunoprecipitates of endogenous c-Myc (Fig. 4).
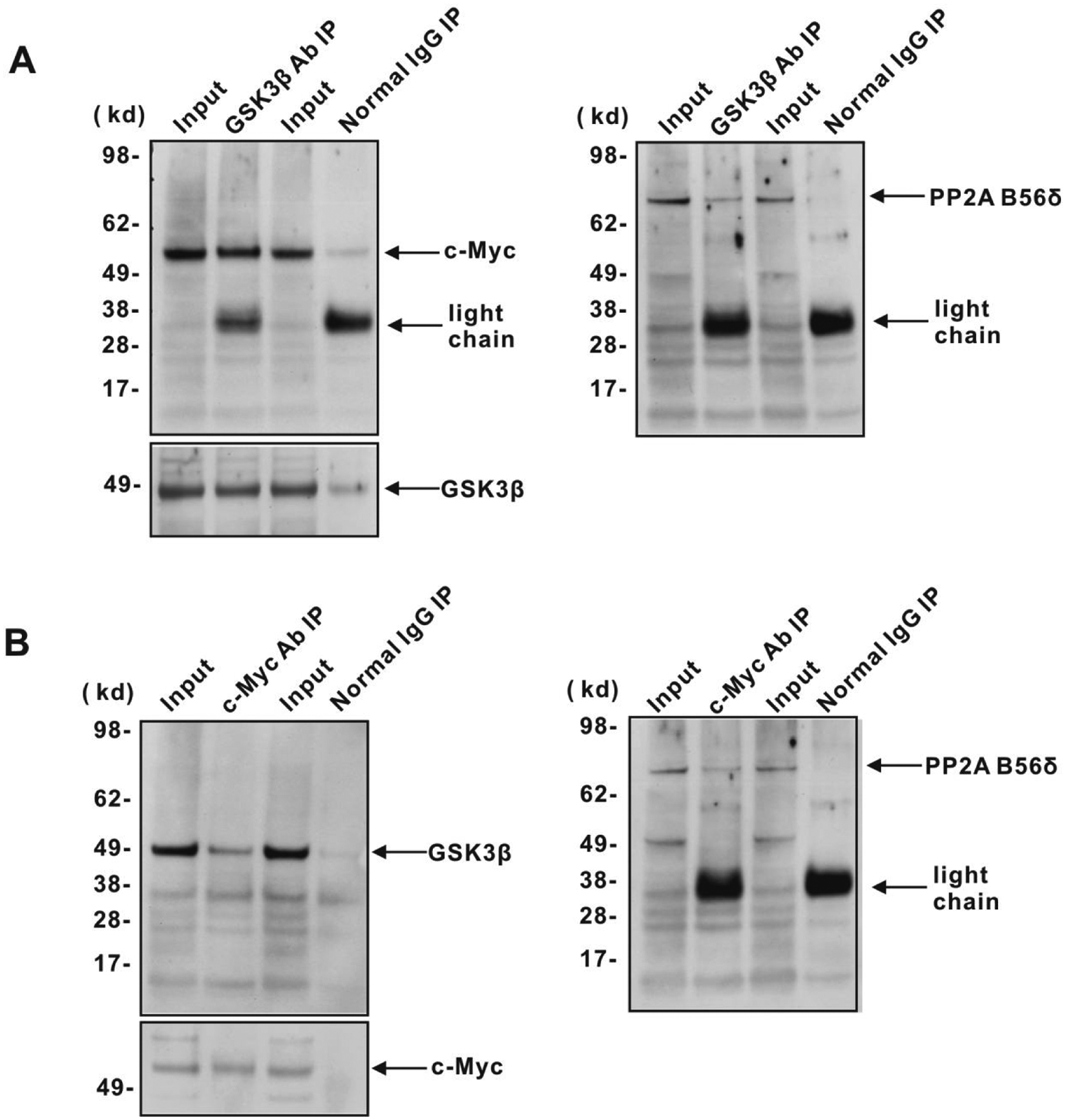
B56δ interacts with GSK3β and c-Myc. Immunoprecipitation and immunoblotting were performed from P493-6 cell lysates to examine the interaction between endogenous c-Myc, GSK3β, and B56δ. Cells were cross-linked by 1% formaldehyde and then lysed for immunoprecipitation with (
To determine whether B56δ levels affected phosphorylation of GSK3β or Myc, we introduced increasing amounts of V5-His-tagged B56δ into 293T cells. As shown in Figure 5A and 5B, we found that increased B56δ resulted in diminished GSK3β S9 phosphorylation without any change in total GSK3β levels. However, c-Myc protein levels were decreased, likely related to the increased phosphorylation of c-Myc T58 and decreased phosphorylation of S62 (Fig. 5A and 5B). These sites are located within the MBI phosphodegron, where PP2A-mediated dephosphorylation of S62 and GSK3β-mediated phosphorylation of T58 are associated with ubiquitination and degradation of Myc. In order to directly examine the effects of PP2A B56 subunits on c-Myc stability, we expressed B56α, B56β, B56γ, and B56δ subunits in 293T cells (Suppl. Fig. S4A) and measured c-Myc protein half-life in cycloheximide chase assays (Suppl. Fig. S4B and Fig. 5C). As shown in Figure 5C, B56α and B56γ produced only a marginal decrease in c-Myc half-life, whereas both B56β and B56δ decreased c-Myc half-life by 50%.
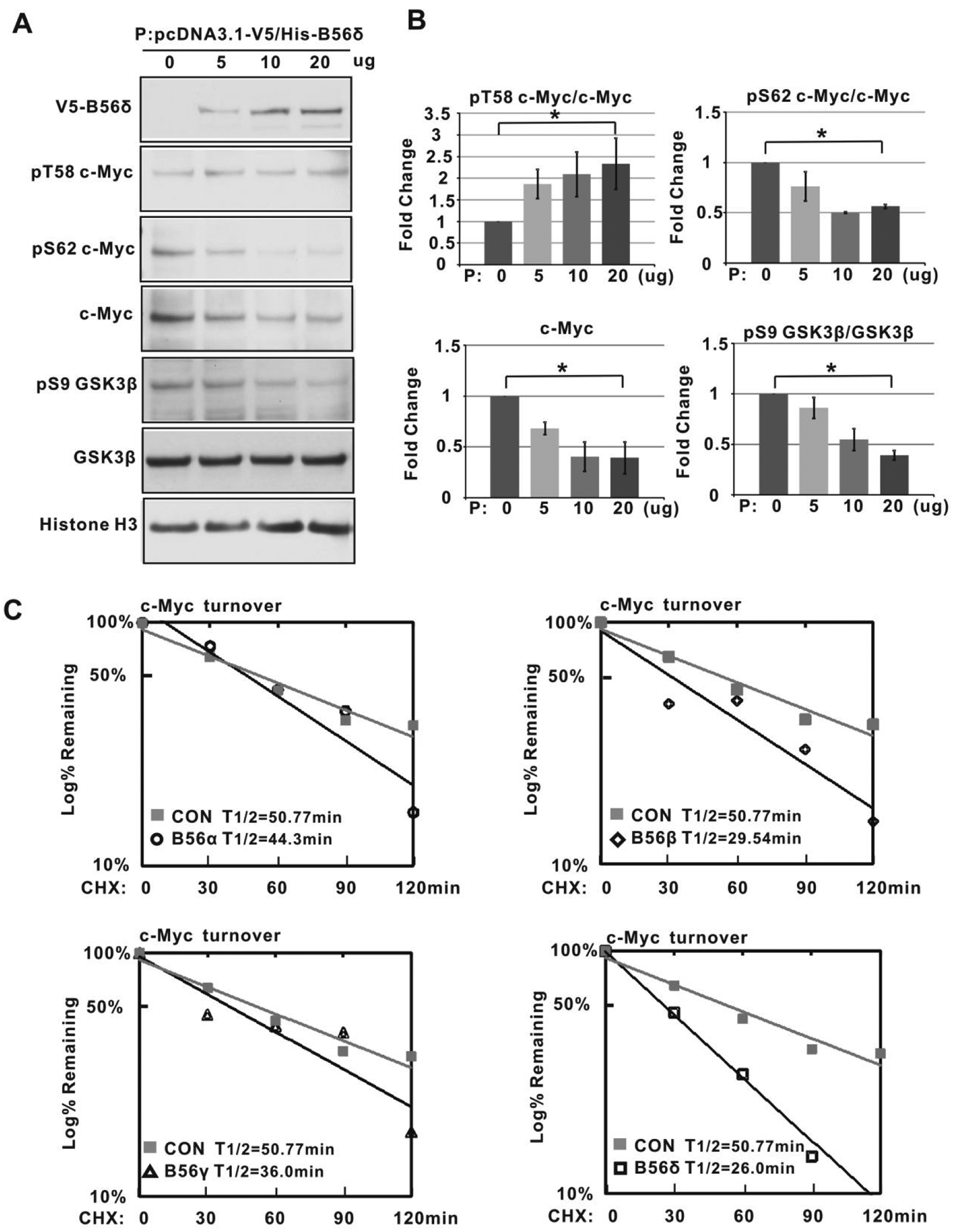
B56δ promotes GSK3β dephosphorylation and c-Myc degradation. (
To further determine whether B56δ is required to maintain the normal half-life of the c-Myc protein, we made B56δ stable knockdown293T cell lines with lentiviral vectors expressing B56δ shRNAs. Compared to control shRNA-expressing cells, the cells with stably knocked down B56δ showed decreased levels of B56δ and increased GSK3β S9 phosphorylation (Fig. 6A). We also found that transient siRNA knockdown of B56δ leads to increased levels of GSK3β phospho-S9 compared to control siRNA-transfected cells (Fig. 6B). We next carried out cycloheximide chase experiments in control, B56δ-overexpressing, and B56δ stably knocked down cells to determine c-Myc protein half-life (Fig. 6C). In this experiment, the c-Myc half-life in control cells was about 45 minutes and was reduced to 30 minutes upon B56δ overexpression (Fig. 6D). By contrast, B56δ knockdown resulted in an increased half-life of 100 minutes (Fig. 6D). To examine the effect of B56δ knockdown in a nontransformed cell line, we introduced B56δ shRNA into human foreskin fibroblasts. Consistent with our observation that B56δ knockdown in 293T cells prolongs c-Myc half-life, we find that in human foreskin fibroblasts, increased levels of endogenous c-Myc protein are evident upon B56δ knockdown (Suppl. Fig. S5).
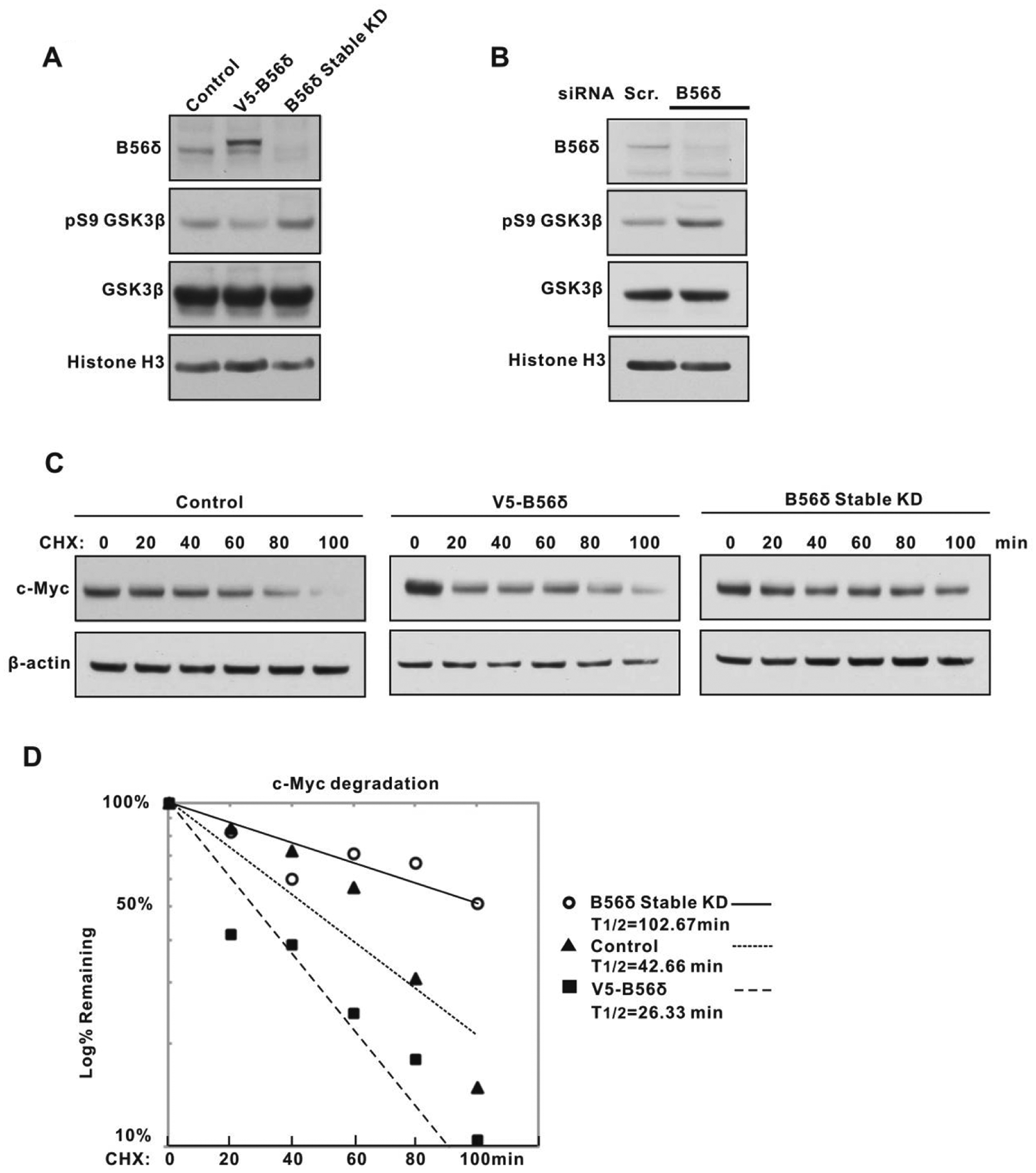
B56δ regulates GSK3β activity and c-Myc degradation. (
Discussion
There is considerable evidence that Myc protein abundance is controlled at multiple levels.5,6 Recently, much interest has focused on the modulation of Myc protein degradation by means of signal transduction pathways that converge on the MBI phosphodegron. 56 Here, we show that Myc participates in the regulation of its own half-life through direct binding and transcriptional activation of the ppp2r5d gene encoding the B56δ regulatory subunit of PP2A (Fig. 3).
PP2A represents a family of heterotrimeric serine-threonine phosphatases whose subunit composition, assembly, and activity are regulated by numerous cellular factors.29,57 Through its B subunits, encoded by at least 15 distinct loci, PP2A achieves a high degree of substrate binding specificity. In particular, the B56 subunits have been shown to be responsible for highly specific PP2A regulation of pathways as diverse as TGF-β and β-catenin signaling and cortical M-type potassium channel activity.49,58,59 PP2A-B56 has also been linked to Myc stability, 30 and its importance is underscored by the finding that CIP2A, an inhibitor of PP2A that is overexpressed in human malignancies, binds to Myc and blocks Myc degradation. 60 A common element in numerous PP2A-B56–regulated pathways is GSK3β, whose inhibitory phosphorylation at S9 by AKT/PKB is reversed by PP2A-B56 phosphatase activity.26,39,40,49,59 We have found that induction of Myc in several cell types (including B lymphoid cells, epithelial cells, and rat and human fibroblasts) correlates with decreased GSK3β S9 phosphorylation concomitant with increased kinase activity (Figs. 1 and 2). This Myc-induced activation of GSK3β occurs in a PP2A-dependent manner because it correlates with increased PP2A activity (Fig. 2C) and is blocked by the PP2A inhibitor OA (Fig. 2A and 2B). Further, we found that while induction of Myc had little, if any, effect on expression of the PP2A catalytic C subunit or the B56α, β, γ, or ϵ subunits, it led to strongly increased expression of the B56δ subunit (Fig. 3A-C). Interestingly, of all the B56 isoforms, only B56δ was reported to direct PP2A to both nuclear and cytoplasmic fractions. 53 In addition, we found that PPP2R3B (PR48) level also increases in Myc-on cells (unpublished data), which may also contribute to Myc-mediated PP2A activation. The B56δ subunit had been previously linked to GSK3β activation by PP2A.49,59 Our expression data as well as the chromatin immunoprecipitation (ChIP) and reporter assays demonstrate that B56δ is a direct transcriptional target of Myc (Fig. 3D-F).
Multiple signaling pathways utilize GSK3β; however, it has been suggested that to some degree, this kinase is “insulated” within each pathway; in other words, distinct pools of GSK3β exist, only a subset of which may be affected by specific signals or modifications. 25 Because GSK3β plays a direct role in Myc stability through its phosphorylation of T58 within the MBI phosphodegron,22,61,62 we asked whether changes in B56δ levels impact the half-life of the Myc protein. We found that increasing the levels of B56δ significantly shortens the Myc protein half-life, while knockdown of B56δ lengthens half-life relative to control cells (Fig. 6). These results support a model, depicted in Figure 7, in which Myc induces expression of B56δ that in turn, as a subunit in a PP2A complex, directs dephosphorylation of GSK3β S9. This leads to activation of GSK3β and phosphorylation of Myc T58, thereby triggering Fbw7 binding and proteasome-mediated degradation. However, in order to phosphorylate Myc T58, GSK3β requires a priming phosphorylation at Myc S62. This requirement would be expected to constrain the action of the Myc-B56δ-GSK3β feedback pathway such that it is dependent on kinase activities that target S62 (e.g., Ras-MAPK, cyclin B/CDK1). This dual-signal input system may help explain why even high levels of Myc appear to maintain or increase protein stability rather than lead to increased degradation through augmented induction of B56δ. Myc stability may also be reinforced through mutations or factors, such as CIP2A, 60 that suppress PP2A and GSK3β activation.
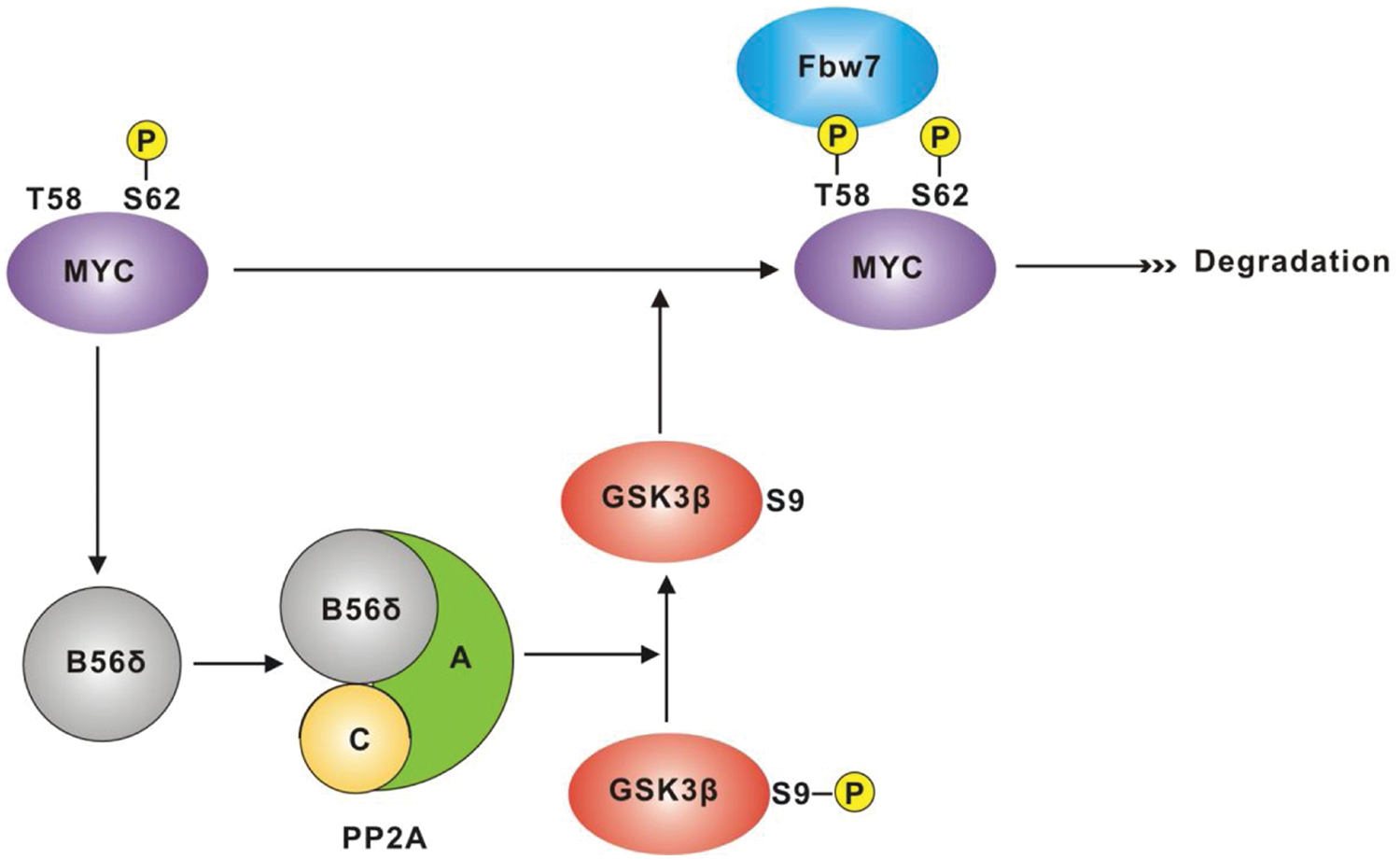
Model of a Myc-B56δ-GSK3β–negative feedback pathway. The diagram depicts Myc induction of B56δ expression (by direct transcriptional activation of the ppp2r5d gene). B56δ is incorporated into a PP2A complex, which in turn mediates dephosphorylation of GSK3β phospho-S9, leading to increased GSK3β kinase activity. Myc phospho-S62 acts as a priming signal, directing active GSK3β to phosphorylate Myc T58. Myc phospho-T58 is then recognized by the ubiquitin ligase Fbw7-SCF complex, leading to proteasome-mediated degradation of Myc. PP2A has also been linked to dephosphorylation of Myc S62.
We observed that B56δ associates with GSK3β (Fig. 4A) and that introduction of increasing amounts of B56δ into 293T cells resulted in heightened levels of Myc T58 phosphorylation along with decreased amounts of Myc protein (Fig. 5A and 5B). These findings can be explained by augmented GSK3β activity due to B56δ-PP2A. However, we also noted that B56δ was present in Myc immunoprecipitates (Fig. 4A) and that B56δ expression correlated with decreased levels of phosphorylated Myc S62 (Fig. 5B). Previous work has established that an Axin-Pin1-GSK3β-PP2A/B56α complex is associated with Myc and is linked to Myc degradation.28,31 Altered levels of complex subunits and PP2A activity have also been tied to Myc stabilization and to clinical outcome in melanomas and breast cancers as well as squamous cell and colon carcinomas.18,60,63-65 Of the 4 B56 subunits, we found that only B56β and B56δ overexpression led to a shorter half-life for Myc, while the B56α and B56γ subunits had relatively smaller effects (Fig. 5C). While our data implicate Myc in controlling expression of B56δ, other pathways are likely to regulate B56β to coordinately control GSK3β activity. 49 Interestingly, changes in single subunits have been shown to permit PP2A to distinguish between immediately adjacent phosphorylation sites in the same protein. 66 It remains to be determined whether PP2A/B56δ is present in a Myc-associated complex that is functionally distinct from the B56α complex previously described.
In summary, our findings establish a role for B56δ as a Myc target and a component of an autoregulatory loop that modulates Myc protein turnover through PP2A-mediated activation of GSK3β (Fig. 7). Because B56δ is also involved in other critical dephosphorylation events (e.g., β-catenin signaling, ion channel activation, activation of G2/M), regulation of B56δ by Myc provides a means through which Myc may exert control over multiple cellular pathways.
Materials and Methods
Plasmids
PPP2R5A, PPP2R5B, PPP2R5C, and PPP2R5D cDNA were obtained from the Dana-Farber/Harvard Cancer Center DNA Resource Core (Boston, MA). Full-length ppp2r5a, ppp2r5b, ppp2r5c, and ppp2r5d were cloned into pcDNA 3.1/V5-His TOPO vector (Invitrogen, Carlsbad, CA). pD40-His/V5-c-Myc was a generous gift from Rosalie C. Sears (Department of Molecular and Medical Genetics, Oregon Health and Science University, Portland, OR).
Antibodies
c-Myc Ab5 (clone 67P05) (Thermo Fisher Scientific, Waltham, MA); purified mouse anti-GSK3β, purified mouse anti-GSK3β (pY216) (BD Biosciences, Franklin Lakes, NJ); phospho-GSK3β (5B3) rabbit mAb, GSK3β (27C10) rabbit mAb, β-actin (13E5) rabbit mAb, anti-Pin1, phospho-(Ser/Thr) AKT substrate antibody, AKT antibody, phospho-AKT (Thr308) (C31E5E) rabbit mAb, phospho-AKT (Ser473) (193H12) rabbit mAb, PP2A C subunit (52F8) rabbit mAb, Mcl-1 antibody (Cell Signaling Technology, Danvers, MA); mouse anti-V5 tag antibody (Invitrogen); monoclonal anti-histone H3, mouse anti–phospho-MBP (clone P12) (Millipore, Billerica, MA); anti–PP2A-B′ (B56) (Millipore); anti-PPP2R5D (Bethyl Laboratories, Montgomery, TX); anti–c-Myc phospho-S62 antibody was a generous gift from Jukka Westermarck (Centre for Biotechnology, University of Turku, Turku, Finland).
In vitro GSK3β kinase assays
In vitro kinase assays were performed with GSK3β antibody using the Non-Radioactive Kinase Assay Kit (Millipore). Briefly, GSK3β was immunoprecipitated from P493-6 cell lysates (each sample contains the same quantity of protein based on BCA assay) using GSK3β rabbit mAb conjugated to Dynabeads Protein G (Invitrogen). ATP (500 uM), dephosphorylated MBP (dp-MBP) (5 ug), and purified GSK3β were added to kinase buffer and incubated at 30°C for 20 minutes. BIO, the specific GSK3β inhibitor, was used to block the activity of GSK3β in the reaction. The reaction was terminated by the addition of 2× LDS sample buffer, electrophoresed using SDS/PAGE, and proteins transferred to nitrocellulose membranes. The phosphorylation state of the MBP was assessed by immunoblotting with anti–phospho-MBP/IR dye 800CW (goat anti-mouse) using the Odyssey Infrared Imaging System (LI-COR, Lincoln, NE) for imaging and quantification.
In vitro PP2A phosphatase activity assessment
PP2A phosphatase activity was measured using a pNPP hydrolysis assay with a Ser/Thr Phosphatase Assay Kit (Millipore) and following the manufacturer’s directions. Determination of PP2A activity was based on reduction of phosphatase activity following treatment with OA (10 nM, 24 hours), a specific inhibitor of PP2A at the concentration used. 41 Briefly, cell lysates containing equal concentrations of protein (20 ug) were incubated with 200 uL of pNPP substrate solution (180 ug) in Ser/Thr assay buffer (containing NiCl2: 1 mM) in 37°C for 2 hours. The reaction was stopped by the addition of 50 uL 13% K2HPO4. Absorbance at 405 nM on a scanning spectrophotometer was used for quantification. All samples were assayed in triplicate, and the experiments were repeated 3 times.
Cell transfections, immunoprecipitation, and immunoblotting
293T cells were transfected with various plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. For co-immunoprecipitation assays employing protein cross-linking, cells were treated with 1% formaldehyde for cross-linking for 10 minutes. 67 The supernatant was removed, and the reaction was quenched with 0.5 mL ice-cold 1.25 M glycine/PBS. Cell lysates were prepared with RIPA buffer and incubated with 2 µg of specified antibody for 2 hours followed by the addition of Dynabeads Protein G (Invitrogen) and shaking for 4 hours at 4°C. The immunocomplexes were washed with RIPA buffer 5 times and applied to SDS-PAGE. Immunoblotting was carried out following standard protocols. For co-immunoprecipitation assays without protein cross-linking, the cells were lysed using low-stringency lysis buffer (150 mM NaCl, 20 mM Tris HCl, 0.1% NP-40, pH 7.4, supplemented with protease and phosphatase inhibitors). The 300 uL cell lysate was precleared with 30 uL Protein G magnetic beads (New England Biolabs, Ipswich, MA) and incubated for 30 minutes at 4°C. Primary antibody was added to the cell lysate and incubated for 2 hours at 4°C, followed by 30 uL Protein G magnetic beads (New England Biolabs), and incubated for another 2 hours. The immunocomplexes were then washed with mild lysis buffer 5 times and applied to SDS-PAGE.
6-His-tag pull-down assay
293T cell lysates were incubated with Dynabeads His-Tag Isolation & Pull Down beads (Invitrogen) for 20 minutes at 4°C using an orbital shaker. After magnetic separation of the beads, they were washed 4 times with 300 uL binding/wash buffer. Hiselution buffer (100 ml was added to the pellet and the mixture placed on a roller for 5 minutes at room temperature. Beads were removed using a magnet and the supernatant containing the eluted protein processed for immunoblotting.
Quantitative RT-PCR
Total RNA was isolated using TRIzol Reagent (Invitrogen) according to the manufacturer’s protocol, and cDNA was transcribed using M-MLV Reverse Transcriptase (Invitrogen) and random hexanucleotide primers. Quantitative RT-PCR analysis was performed in triplicate using SYBR Green qPCR superMix-UDG (Invitrogen) assayed on an ABI 7900HT Real Time PCR System (Applied Biosystems, Foster City, CA). Data were calculated according to the ΔΔCt relative quantification method. Sequences of primers used to amplify the target genes are provided in Supplementary Table S1.
ChIP and real-time PCR
Cells were cross-linked with formaldehyde as described above, and chromatin was immunoprecipitated following the cross-linking chromatin immunoprecipitation (X-ChIP) protocol (Abcam, Cambridge, UK). Anti–c-Myc antibody N262 was used to immunoprecipitate chromatin fragments using ChIP-Grade Protein G Magnetic Beads (Cell Signaling Technology). Real-time PCR was performed using an ABI 7900 Sequence Detection System (Applied Biosystems) with SYBR Green qPCR superMix-UDG (Invitrogen). Sequences of primers used to amplify ChIP samples are provided in Supplementary Table S1.
RNA interference
siRNAs against c-Myc (FlexiTube siRNA no. SI00300902) and B56δ (FlexiTube siRNA no. SI02653350) were purchased from Qiagen (Hilden, Germany). Transfection was carried out twice with a 24-hour interval with 100 nM siRNA using Lipofectamine RNAiMAX reagent according to the manufacturer’s protocol (Invitrogen). At 48 hours after transfection, the cells were harvested for detection.
Cycloheximide chase assay
Cells were treated with 100 ug/mL cycloheximide, harvested at the indicated time points, and subjected to immunoblotting.
Lentivirus knockdown
The pGIPZvector with nonsilencing control shRNA and vectors containing shRNA against PPP2R5D (V2LHS_57934 and V2LHS_57937) were purchased from Open Biosystems (Huntsville, AL). Fifteen micrograms of pGIPZ vector, together with 7.5 µg of each packaging vector (pMD2.G and psPAX2), were co-transfected into 293T cells. Supernatant-containing lentivirus particles were harvested 48 hours and 72 hours after transfection, passed through a 0.45-µm membrane filter, and directly used to infect prostate cancer cells in the presence of 6 µg/mL polybrene. After 48 hours, the infected cells were maintained in the medium with 2 µg/mL puromycin for 1 week before knockdown assessment.
Cloning and mutation of ppp2r5d (B56δ) promoter and luciferase reporter assay
Genomic DNA was extracted from P493-6 cells using a salt extraction method. 68 The ppp2r5d (B56δ) promoter sequence (2.4 kb) was amplified from genomic DNA using primers shown in Supplementary Table S1 (B56δproKpnI-F/B56δproXhoI-R) and subcloned into pGL4.70 (Promega, Fitchburg, WI). Two E-boxes in the B56δ promoter were mutated using overlapping extension PCR 69 employing the primers in Supplementary Table S1 (B56δproKpnI-F/B56δpro_EM_LR; B56δpro_EM_ LF/B56δproXhoI-R) and then cloned into pGL4.70 (Promega).
An HFF cell line stably expressing Myc (infected with pBABE–c-Myc) and an HFF control cell line (infected with pBABE alone) were transfected with pGL4.70-WT B56δpro or pGL4.70-MUTB56δpro. At 24 hours after transfection, the cells were harvested and submitted to luciferase reporter assay using the Dual-Luciferase Reporter Assay Kit (Promega) and read by TopCount NXT Microplate Scintillation and Luminescence Counter (PerkinElmer, Waltham, MA).
Footnotes
Acknowledgements
We are grateful to Rosalie Sears (Oregon Health and Science University) and Jukka Westermarck (Centre for Biotechnology, University of Turku, and Åbo Akademi University) for kind gifts of reagents. We also thank Daniel Diolaiti and Maralice Conacci-Sorrell for critical readings of the article and the members of the Eisenman Laboratory for discussions and advice throughout the course of this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health, National Cancer Institute (R37-CA57138 to R.N.E.), and a Doug & Maggie Walker Postdoctoral Fellowship (to L.L.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.