
Abstract
Mitotic division is induced by protein phosphorylation. For a long time the supported hypothesis was that mitotic entry and exit were the exclusive result of cyclin B-Cdk1 kinase activation and inactivation, whereas the phosphatase activity required to dephosphorylate mitotic substrates was thought to be constant during mitosis. Recent data demonstrate that phosphatase activity must also be tightly regulated to promote correct cell division. Here we describe the new pathway involved in phosphatase regulation and the questions that this discovery raises concerning the classic view of cell cycle regulation.
Introduction
Mitotic entry is driven by the activation of the cyclin B-Cdk1 complex. 1 This kinase promotes a broad range of protein phosphorylation that results in ordered structural rearrangements in the cell, such as nuclear envelope breakdown, chromosome condensation, and spindle formation. Cyclin B-Cdk1 is activated at the G2-M transition through the dephosphorylation of the inhibitory residues of Cdk1 by the dual phosphatase Cdc25.2-4 Mitotic progression is then mediated by the activation of the ubiquitin ligase anaphase promoting complex (APC) at the metaphase-anaphase transition. This complex promotes securin and cyclin B ubiquitination and degradation, resulting in chromosome segregation and cyclin B/Cdk1 inactivation. 5 The majority of mitotic phosphorylations are then removed, and mitotic exit takes place. Activation and inactivation of cyclin B-Cdk1 complex have been thought to be the key events promoting mitotic entry and exit. However, it is obvious that phosphatases are required to hydrolyze the serine/threonine phosphoesters induced by this mitotic kinase. 6 For a long time, the idea that phosphatase activity was constant during mitosis was supported; however, recent findings indicate that as for the mitotic kinase, the activity of phosphatases must be tightly regulated to promote normal cell division. Thus, mitotic division is the result of a subtle balance between cyclin B-Cdk1 and its counteracting phosphatases that finely regulate protein phosphorylation.
The Gwl-PP2A Pathway
The implication of phosphatases in mitotic entry and exit in higher eukaryotes has been much studied in the last 20 years, mostly by the use of phosphatase inhibitors.7,8 However, in 2009, two independent laboratories simultaneously identified the phosphatase involved in cyclin B-Cdk1-substrate dephosphorylation during mitosis.9,10
The first observations from one of these laboratories showed that a high phosphatase activity opposed to mitotic phosphorylation was present in interphase Xenopus egg extracts. 10 This activity was sensitive to 200 nM okadaic acid, suggesting that it could be due to PP2A. PP2A phosphatase is a complex of a catalytic C-subunit, a noncatalytic scaffolding A subunit, and a third variable B-subunit that acts as a substrate specifier. These B subunits have been classified into 4 types: B (B55), B′ (B56), B′′ (PR72), and B′′′ (PR93), and some of these B subunits are coded by several related genes resulting in different isoforms (α, β, γ, and δ for B55 and α, β, γ, δ, and ϵ for B56). 11 The combination of all these subunits results in the formation of more than 200 distinct holoenzymes. By immunodepletion of the different B subunits of PP2A in Xenopus egg extracts, the authors of this first study demonstrated that PP2A-B55δ was the phosphatase whose activity counteracted cyclin B-Cdk1 protein phosphorylation during mitosis. 10
The second laboratory focused its work in the role of the Greatwall (Gwl) kinase in mitosis. Gwl was originally identified in Drosophila, where it was first proposed to be involved in the control of mitotic progression.12-14 Further studies in Xenopus egg extracts suggested that its activity was required to maintain metaphase II arrest by preventing cyclin B-Cdk1 inactivation. 15 However, the authors of this second study demonstrated that co-depletion of Gwl from mitotic egg extracts with the inhibitory kinases of cyclin B-Cdk1, Myt1 and Wee1, still induced mitotic exit. These results indicated that Gwl is required to maintain the mitotic state even in the presence of high cyclin B-Cdk1 activity and suggested that this kinase was inhibiting a phosphatase involved in cyclin B-Cdk1 substrate dephosphorylation. 9 Finally, in this study, mitotic exit promoted by Gwl, Myt1, and Wee1 elimination in mitotic egg extracts was rescued by immunodepletion of PP2A, demonstrating that PP2A was the phosphatase regulated by Gwl during mitosis (Fig. 1). All these data were subsequently confirmed in 3 other studies developed in Xenopus egg extract and Drosophila models.13,16,17
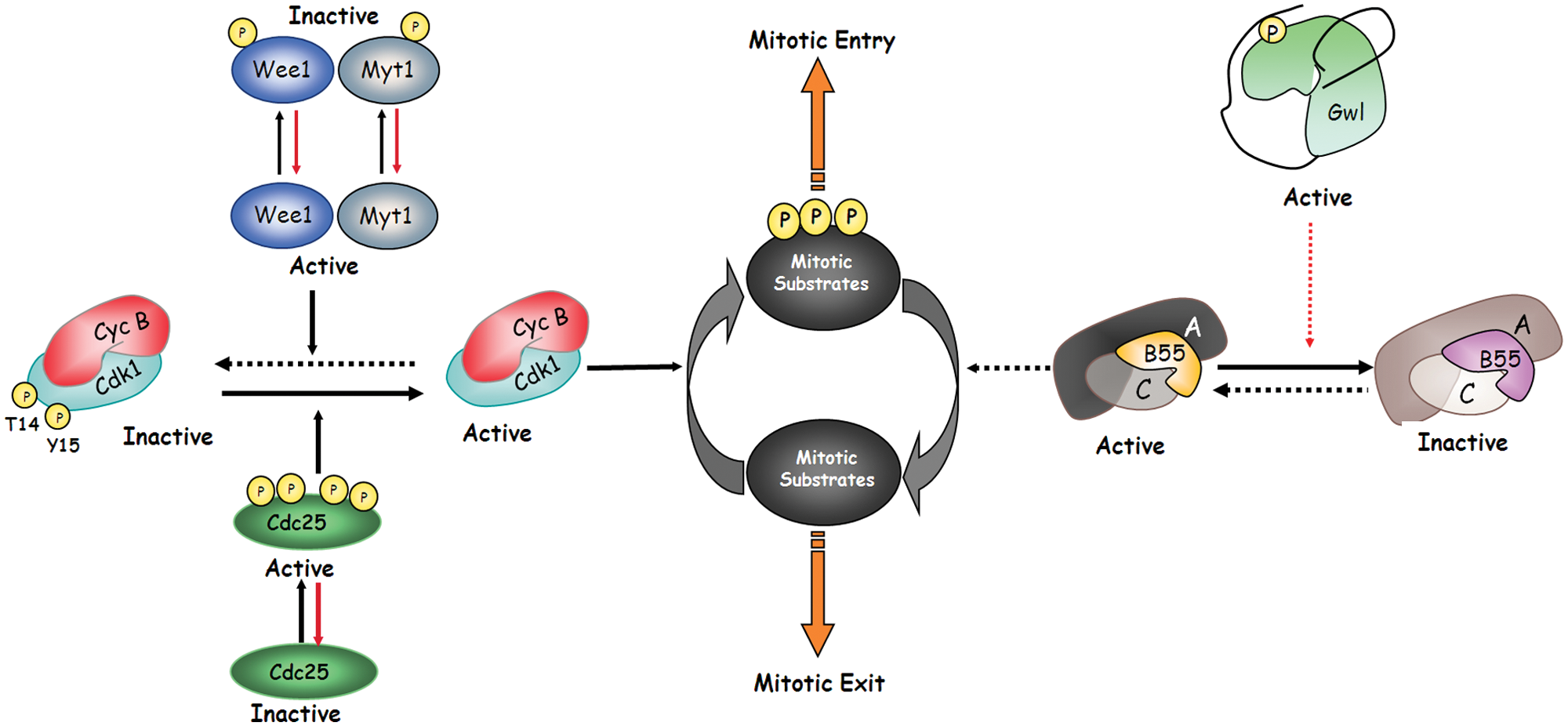
Two kinases are required to promote mitotic entry. Cyclin B-Cdk1 and Gwl are both required for mitotic entry. Cyclin B-Cdk1 is activated at G2-M by dephosphorylation of the inhibitory residues of Cdk1 threonine 14 and tyrosine 15 by the dual phosphatase Cdc25. Once activated, cyclin B-Cdk1 promotes phosphorylation of mitotic substrates. In contrast, Gwl activation at mitotic entry promotes the inhibition of PP2A-B55, the phosphatase responsible for mitotic substrate dephosphorylation. Dotted lines represent inactive pathways during mitosis. Dotted red lines symbolize indirect regulation.
Arpp19 and ENSA, Two First Substrates of Gwl Inhibiting PP2A-B55
Since data from several laboratories suggested that Gwl did not directly regulate PP2A-B55 activity, identification of the substrates of Gwl mediating this inhibition during mitosis was essential. The same 2 laboratories involved in PP2A identification independently discovered the c-AMP regulated phosphoprotein 19 (Arpp19) as the first substrate of Gwl. Both studies used classic biochemical purification in Xenopus egg extracts and in vitro phosphorylation assays to obtain a phosphorylated band of 19 KDa.18,19 Among the mass spectrometry sequenced proteins in this band, Arpp19 was identified as the Gwl substrate. Arpp19 is a cAMP-regulated phosphoprotein first found in mammalian brain as an in vitro substrate of protein kinase A (PKA). This protein has been suggested to be involved in neuronal development and regeneration through the stabilization of the mRNA for growth-associated protein 43 (GAP-43). 20 Arpp19 is highly homologous to another small phosphoprotein, α-endosulfine (ENSA). ENSA was originally identified as a possible endogenous ligand for the sulfonylurea binding site of KATP channels; however, its cytosolic localization makes it unlikely that this protein plays such a role in vivo. 21 However, evidence for a role of endos, the unique gene of the Arpp19/ENSA family in Drosophila, in cell cycle progression was described. Oocytes from Drosophila deficient for endos do not progress into meiosis. 22 They present a high cyclin B-Cdk1 activity but low phosphorylation of the substrates of this kinase, a situation that mimicked the one observed in mitotic Xenopus egg substrates in which Wee1/Myt1 and Gwl were immunodepleted. 9
Human purified Arpp19 and ENSA proteins are phosphorylated in vitro by Gwl (S62 and S67, respectively), and when added to interphase Xenopus egg extracts, this phosphorylation promotes the binding to and inhibition of the PP2A-B55 holocomplex, resulting in mitotic entry.18,19 However, despite the fact that both ectopic proteins can promote entry into mitosis, the exact role of the endogenous proteins in mitotic division is much less clear. In this regard, in one of the studies immunodepletion of ENSA did not affect the mitotic state, whereas Arpp19 prevented mitotic entry or promoted mitotic exit when immunodepleted from interphase or mitotic egg extracts, respectively. 19 In contrast, in the second study, immunodepletion of ENSA from cycling extracts prevented PP2A-B55 inactivation and phosphorylation of mitotic substrates in the presence of high cyclin B-Cdk1 activity. These puzzling results could be explained either by the different egg extracts used in the experiment (mitotic/interphase versus cycling extracts) or by a different capacity or specificity of the antibodies used for immunodepletion. Further studies are required to clarify the exact contribution of these 2 proteins in mitotic division.
Besides Gwl-dependent phosphorylation, Arpp19 and ENSA are also phosphorylated by PKA (S104 and S109) and by Cdk1. 18 The role of these phosphorylations is unknown. Our unpublished results suggest that they are not essential for phosphatase binding; however, they could participate in the fine modulation of PP2A to confer the correct timing of inhibition under different in vivo conditions.
Of particular interest is the role of PKA in prophase I arrest of female oocytes. PKA activity plays a major function in prophase arrest of female oocytes during meiosis. In these oocytes, cyclin B-Cdk1 complex is maintained inhibited by the phosphorylation of Cdk1 inhibitory residues. Progesterone promotes a first decrease of PKA activity that is followed by the activation of cyclin B-Cdk1 and meiotic resumption (for review see Perdiguero & Nebreda 23 ). The PKA substrate whose dephosphorylation triggers cyclin B-Cdk1 activation and meiotic progression is unknown. The newly reported function of the PKA phosphoprotein Arpp19 in mediating mitotic entry raises the possibility of a putative role of this protein in meiotic resumption. In this regard, phosphorylation of Arpp19 by PKA in prophase oocytes could modulate Arpp19 phosphorylation by Gwl or decrease the binding affinity of this protein toward the phosphatase. This would ensure a high PP2A-B55 activity and dephosphorylation of cyclin B-Cdk1 substrates during this arrest. Inhibition of PKA by progesterone would restore binding affinity of Arpp19 toward PP2A-B55 and permit cyclin B-Cdk1 activation and meiotic resumption. A similar regulation could be true for mitotic division of mammalian cells that present a PKA activity that oscillates during cell cycle and in which the inhibition of PKA arrests cells in mitosis with high cyclin B-Cdk1 activity.
Gwl Is an Unconventional AGC Kinase with Specific Mechanisms of Activation
Gwl activation is essential to promote mitotic entry. The increase of Gwl activity at G2-M induces PP2A-B55 inhibition, cyclin B-Cdk1 activation, and mitotic entry. Interestingly, Gwl sequence displays a feature that makes this kinase unique and different from all the other known kinases. 24 An insertion of 550 amino acids is present between kinase subdomains VII and VIII at the corresponding T-loop site. 12 Phosphorylation of the T-loop is required for the activation of most kinases; however, the usual length of this domain is about 20 to 30 amino acids, raising the question whether the insert of 550 amino acids in the Gwl kinase can really act as a T-loop. By sequence homology, Gwl belongs to the AGC family of kinases. The mechanism of activation of these kinases involves phosphorylation at 3 different sites: the T-loop, the turn motif present at the C-terminus, and the hydrophobic motif (HM) present at the most C-terminal tail. Stabilization of the active form of the AGC kinases is promoted by the binding of the phosphorylated hydrophobic and turn motifs with 2 different domains at the N-terminus, the hydrophobic pocket and the turn motif–binding site, respectively. Gwl contains a functional hydrophobic pocket and turn motif–binding site at its N-terminus; however, surprisingly, although the turn motif has been identified (S875 in humans, S883 in Xenopus), it is devoid of the HM, a domain that is essential for the rest of the members of the AGC family of kinases. 25
The first work on the study of the mechanisms conferring Gwl activity used a combined phosphomapping and mutagenesis analysis to identify the different phosphorylation sites involved in Gwl activation. Data from this work show that the turn motif site is the unique phosphosite essential for Gwl activation. 25 These data also show that turn motif phosphorylation can be induced in vitro by Cyclin B-Cdk1. However, despite the fact that the in vitro phosphorylation of the turn motif by this kinase promotes activation of a human recombinant Gwl protein, this activity can be increased at least 3 times when the in vitro phosphorylated protein is mixed with a peptide containing the phosphorylated HM of the AGC kinase Rsk2. PDK is the only member of the AGC kinases devoid of HM. The activation of this kinase is mediated by its binding to the phosphorylated HM of other AGC kinases. Authors hypothesize that in a first step, Gwl could be phosphorylated at the turn motif, promoting the binding of the turn motif with the turn motif–binding site and stabilizing the kinase in a partial active form. In a second step, this partial active form would bind to the phosphorylated HM of another AGC kinase, promoting the complete activation of Gwl.
The results of this work indicated that the phosphorylation sites identified in the central insert (putative T-loop) were not essential for Gwl functionality, suggesting that the phosphorylation of the T-loop would not participate in kinase activation. However, a minimal length of this sequence was required for the kinase to have normal activity, suggesting a structural role of this insert.
These results partially differ from those obtained in other laboratory suggesting a function in Gwl activation of 2 phosphosites in the T-loop. 26 Mass spectrometry data identified T193 and T206 (Xenopus sequence) as 2 phosphorylated sites during mitosis. These sites were phosphorylated in vitro by Cdk1 but not by Plx1 and were essential for the subsequent autophosphorylation of the turn motif (S883, Xenopus sequence). From these results, authors suggested that Gwl activation is induced by phosphorylation of T193 and T206 by Cdk1 kinase. This phosphorylation would promote a partial activation of Gwl that would result in the autophosphorylation of the turn motif site and in its complete activation. However, surprisingly, the double phosphomimetic mutant T193E and S883D presents only partial activity. Along this line, to clarify the exact role of these phosphorylations on Gwl activation, it would be interesting to determine whether a kinase dead mutant of Gwl can be phosphorylated in the turn motif site when supplemented with mitotic egg extracts.
PP2A-B55/Cyclin B/Cdk1 Balance at Mitotic Entry
At G2 phase, cyclin B-Cdk1 is maintained in an inactive state by phosphorylation of the inhibitory residues of Cdk1 threonine 14 and tyrosine 15 by the inhibitory kinases Myt1 and Wee1.2,3 Conversely, PP2A-B55, the phosphatase that dephosphorylates cyclin B-Cdk1 substrates at its consensus site, is fully active. At the G2-M transition, the dual phosphatase Cdc25 is partially activated and this results in the activation of a pool of cyclin B-Cdk1 by dephosphorylation of the inhibitory residues of Cdk1. This pool of active cyclin B-Cdk1 will now phosphorylate Wee1, Myt1, and Cdc25, promoting the complete inactivation of the inhibitory kinases and activation of the activatory phosphatase. This results in full cyclin B-Cdk1 activity and mitotic entry.27-29 This feedback mechanism of cyclin B-Cdk1 toward its inhibitory kinases and activatory phosphatase is named the cyclin B-Cdk1 activation loop (Fig. 2). However, Wee1, Myt1, and Cdc25—as substrates of cyclin B-Cdk1—are themselves targeted by PP2A-B55, and thus cyclin B-Cdk1 activation cannot take place if this phosphatase is still active.9,15 PP2A-B55 inactivation is mediated by Gwl-dependent phosphorylation of Arpp19. Once phosphorylated, Arpp19 binds and inhibits PP2A-B55. It is only at this moment that Cdc25 will be able to trigger the cyclin B-Cdk1 activation loop. Thus, mitotic entry is triggered by Gwl activation. 30
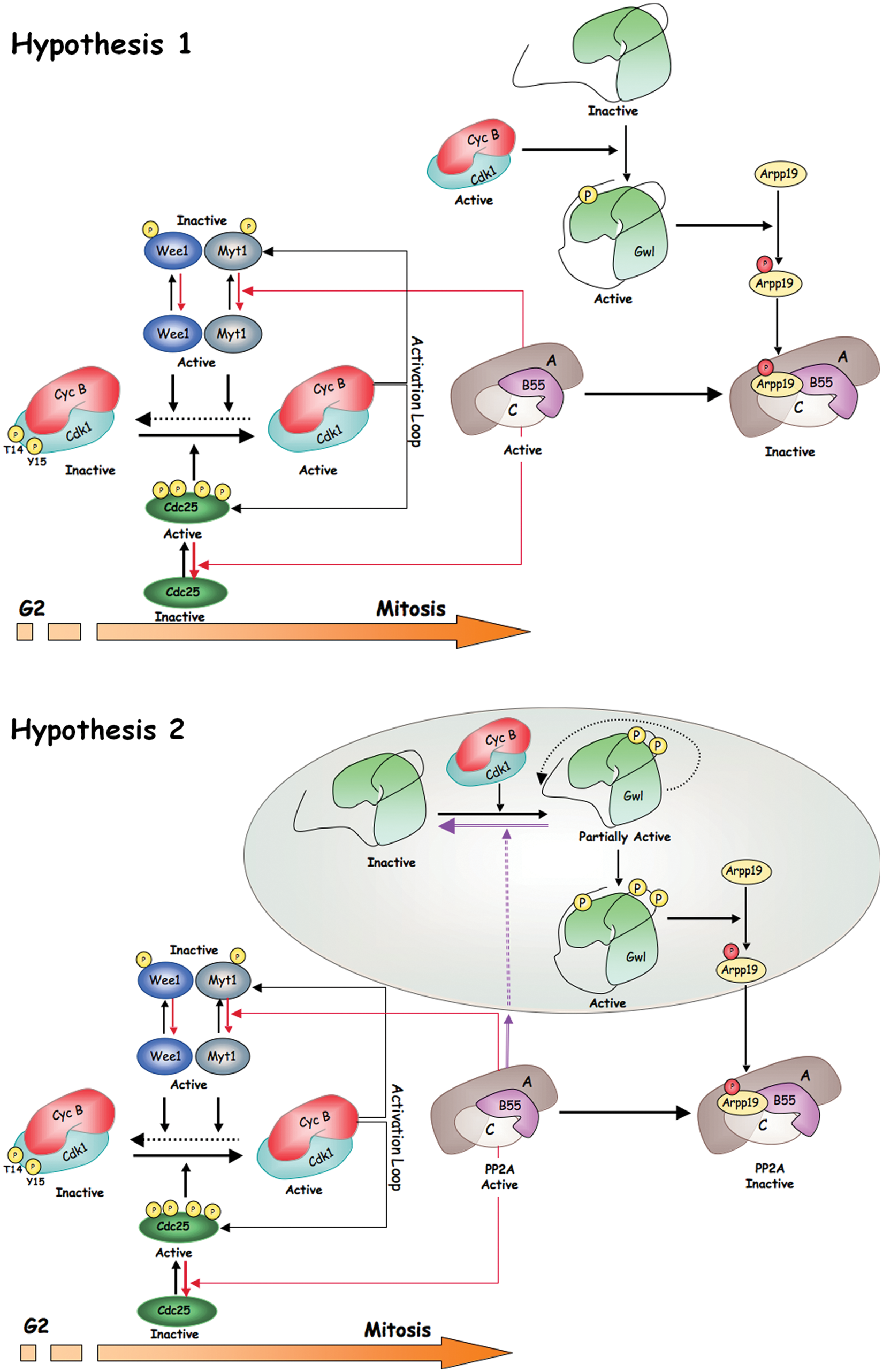
Two hypotheses for Gwl activation and mitotic entry. Two mechanisms of activation of Gwl have been proposed. The first one, represented in Hypothesis 1, involves the sole phosphorylation of the turn motif of Gwl by cyclin B-Cdk1 in a sequence that does not correspond to a cyclin B-Cdk1 consensus site. This phosphorylation would allow the complete activation of Gwl and the subsequent phosphorylation of Arpp19 even in the presence of high PP2A-B55 activity. Phosphorylation of Arpp19 would result in PP2A-B55 inhibition and full phosphorylation of mitotic substrates. In Hypothesis 2, Gwl would be first activated by phosphorylation of this protein in 2 S/T-P sites of the T-loop by cyclin B-Cdk1 complex. This would promote the autophosphorylation of the turn motif site of the Gwl kinase and its complete activation. Activated Gwl would phosphorylate Arpp19 that would subsequently inhibit PP2A-B55. In this hypothesis, Gwl/Arpp19 would be localized far from PP2A-B55 to prevent dephosphorylation of the T-loop sites of Gwl and to permit the complete activation of this kinase. Colored circle represents a distinct cellular localization.
One of the current hypotheses proposes that Gwl activation at G2 results from the phosphorylation of this kinase by cyclin B-Cdk1. Taking into account the data reported above on the mechanisms promoting Gwl activation, we could imagine that Gwl activation requires the sole phosphorylation of the turn motif by cyclin B-Cdk1. 25 The turn motif is not a cyclin B-Cdk1 consensus site, and consequently it is likely not be a substrate of PP2A-B55 during G2. Therefore, only a minimal phosphorylation of Gwl in this site would be sufficient to trigger a partial phosphorylation of Arpp19 and inhibition of PP2A-B55, resulting in the final activation of cyclin B-Cdk1 and mitotic entry. Along this line, it is possible that the basal activity of cyclin A-Cdk1 plus cyclin B-Cdk1 that increases through the G2 phase could reach a level sufficient to promote the phosphorylation of a partial pool of Gwl. Since this phosphorylation at the turn motif does not correspond to the cyclin B-Cdk1 consensus site, it will not be dephosphorylated by PP2A-B55 and will act as the trigger event promoting mitotic entry (Fig. 2, Hypothesis 1).
In an alternative hypothesis, Gwl would reach complete activity by a first phosphorylation of this kinase in the 2 cyclin B-Cdk1 consensus sites of the T-loop followed by a subsequent autophosphorylation of the turn motif. 26 This hypothesis would imply that Gwl and Arpp19 would co-localize during G2 in the same subcellular structure, far from PP2A-B55. After Arpp19 phosphorylation by Gwl, this protein would be transported to a new subcellular localization where it would bind and inhibit PP2A-B55, allowing the triggering of the cyclin B-Cdk1 activation loop and promoting mitotic entry (Fig. 2, Hypothesis 2).
Role of the Gwl/Arpp19/PP2A-B55 Pathway at Meiotic Entry and Progression
As for mitosis, the Gwl/Arpp19/PP2A-B55 pathway participates in meiotic division. In Drosophila, a role of endos in mitotic progression has been demonstrated. 22 Data obtained in this model show that endos mutant oocytes arrest at prophase I with a high cyclin B-Cdk1 activity and low phosphorylation of their substrates, indicating that this protein is required to permit cyclin B-Cdk1 substrate phosphorylation during meiosis. Along the same line, overexpression of Gwl and reduction of the Tws protein (the homologue of PP2A-C subunit in Drosophila) induced an arrest of oocytes in meiosis I, suggesting that Gwl promotes meiotic arrest by antagonizing PP2A-B55 activity. 16
In Xenopus oocytes, microinjection of the Gwl protein promotes meiotic resumption, cyclin B-Cdk1 activation, and nuclear envelope breakdown, indicating that this pathway is functional during the first meiotic division. 15 Moreover, despite the fact that there are no data about the role of Gwl in meiosis I to meiosis II progression, it is likely that it could equally participate in this transition. The role of Gwl to promote meiotic division is likely mediated by its capacity to prevent PP2A-B55-dependent dephosphorylation.
Xenopus oocytes are arrested at the prophase of the first meiotic division. The addition of progesterone induces cyclin B-Cdk1 activation, nuclear envelope breakdown, and progression to metaphase I. Metaphase I-II transition is then promoted by the continuous decrease of cyclin B-Cdk1 activity, resulting from a partial degradation of cyclin B. Degradation is not complete since a residual activity of this kinase is essential to prevent DNA replication between metaphase I and II. Finally, the activity of cyclin B-Cdk1 increases again due to cyclin B neosynthesis and oocyte arrest at metaphase II, with high cyclin B-Cdk1 activity and phosphorylated mitotic substrates waiting for fertilization.23,31
The newly identified role of the Gwl-Arpp19-PP2A-B55 pathway in regulation of the phosphorylation of cyclin B-Cdk1 substrates raises a considerable number of questions about how meiotic progression takes place. Now it is important to identify the mechanism by which progesterone promotes cyclin B-Cdk1 activation. In this regard, it is obvious that the activation of cyclin B-Cdk1 after progesterone addition will never happen if PP2A-B55 is not turned off. As reported above, prophase-arrested oocytes present a high PKA activity that fully decreases after progesterone treatment. Since Arpp19 is a substrate of PKA, it is possible that dephosphorylation of Arpp19 at the PKA site could be involved in PP2A-B55 inhibition and cyclin B-Cdk1 activation at meiotic resumption.
Another important question concerns the mechanisms regulating PP2A-B55 activity at the metaphase I-II progression. A partial dephosphorylation of cyclin B-Cdk1 substrates is required for the first polar body emission at meiosis I. This dephosphorylation is the result of a decrease of cyclin B-Cdk1 activity; however, it is obvious that a reactivation of PP2A-B55 is also required. It would be interesting to investigate whether a partial dephosphorylation of Gwl due to a decreased cyclin B-Cdk1 activity would be sufficient to promote partial PP2A-B55 reactivation at meiotic I exit. Similarly, it would be interesting to determine whether the high phosphorylation levels of mitotic substrates required for metaphase II arrest are the result of a maintained and stable inhibition of the PP2A-B55 trough a continuous phosphorylation of Gwl by active cyclin B-Cdk1.
PP2A-B55 Activity Is Required for Mitotic Exit
Cyclin B-Cdk1 activity drives mitosis as far as metaphase, but progression into later phases depends on 2 different mechanisms: first, the ubiquitination and degradation of cyclin B, which in turn inactivates Cdk1; and second, the dephosphorylation of Cdk1 mitotic substrates by phosphatases. 6 This dephosphorylation must be highly coordinated to ensure that cytokinesis does not occur before chromosome segregation. In yeast, protein dephosphorylation at mitotic exit depends on the balance between cyclin B-Cdk1 activity and the phosphatase Cdc14. 32 Much less is known about the phosphatase implicated in mitotic exit in higher eukaryotes. Initial data showing cytokinesis defects due to decreased cyclin B-Cdk1 substrate dephosphorylation in Gwl knockdown human cells suggested a role of the phosphatase PP2A-B55 in this dephosporylation.33,34 This hypothesis was subsequently supported by data obtained in a live-cell imaging RNAi screen in which the B55 subunit of PP2A was identified as a key factor in mitotic exit. 35 The knockdown of this protein promoted defects in spindle breakdown, postmitotic reassembly of nuclear envelope, and chromatin decondensation that correlated with a decreased general phosphorylation of cyclin B-Cdk1 substrates after cyclin B-Cdk1 inactivation. A similar phenotype was reported in Cdc20 knockout cells in which mitotic exit induced by Cdk1 and Greatwall inactivation (named MASTL in humans) was perturbed by B55 knockdown. 36
Apart from PP2A-B55, PP1 phosphatase also appears to participate in protein dephosphorylation in mitotic exit. 37 Accordingly, depletion of this phosphatase from mitotic Xenopus egg extracts prevents cyclin B-Cdk1 substrate dephosphorylation.
However, so far we do not know whether PP2A-B55 and PP1 participate in the direct dephosphorylation of mitotic substrates or whether they participate in this dephosphorylation indirectly through either the activation of other phosphatase or the inactivation of Gwl or/and Arpp19. To clarify this point, it is essential to identify the phosphatases involved in Gwl and Arpp19 dephosphorylation after cyclin B-Cdk1 inactivation.
Metaphase II Arrest Requires a Tightly Regulated PP2A-B55/PP2A-B56 Balance
In vertebrates, unfertilized eggs are arrested in metaphase II of meiosis. This arrest is caused by a cytoplasmic activity named the cytostatic factor (CSF). CSF inhibits the APC, thereby preventing cyclin B degradation and exit of meiosis (reviewed in Lorca & Castro 38 ). The CSF is constituted by the Mos-MAPK-Rsk pathway and the APC inhibitor Emi2. Mos and Emi2 are synthesized during oocyte maturation and are degraded after fertilization. The association of Emi2 with the APC and its inhibition are regulated by multiple phosphorylations of the former protein by different kinases such as Plx1, cyclin B/Cdk1, CK1, and Rsk (Fig. 3). Phosphorylations mediated by cyclin B-Cdk1, Plx1, and CK1 prevent Emi2 from binding the APC, whereas phosphorylation of Emi2 by Rsk stimulates the APC-inhibitory activity of this protein. Interestingly, phosphorylation of Emi2 by Rsk recruits the phosphatase PP2A/B56 β/ϵ, which continuously antagonizes the inhibitory phosphorylations performed by Cdk1/Plx1/CK1. 39 Inhibited APC maintains cyclin B-Cdk1 activity, which in turn will permit the activation of the Gwl-Arpp19 pathway and the subsequent inhibition of PP2A-B55, preventing this massive protein dephosphorylation. It is worth noticing that recruitment and phosphorylation of Emi2 by Plx1 and CK1 depend on the phosphorylation of this protein by cyclin B-Cdk1 in a cluster of consensus sites (S/T-P) and that these sites are recognized by PP2A-B56β/ϵ; however, this phosphate does not massively dephosphorylate cyclin B-Cdk1 substrates. Conversely, PP2A-B55 promotes general dephosphorylation of cyclin B-Cdk1 sites, suggesting that substrate recognition by PP2A-B56 might imply other domains of the protein in addition to the S/T-P consensus sequence.
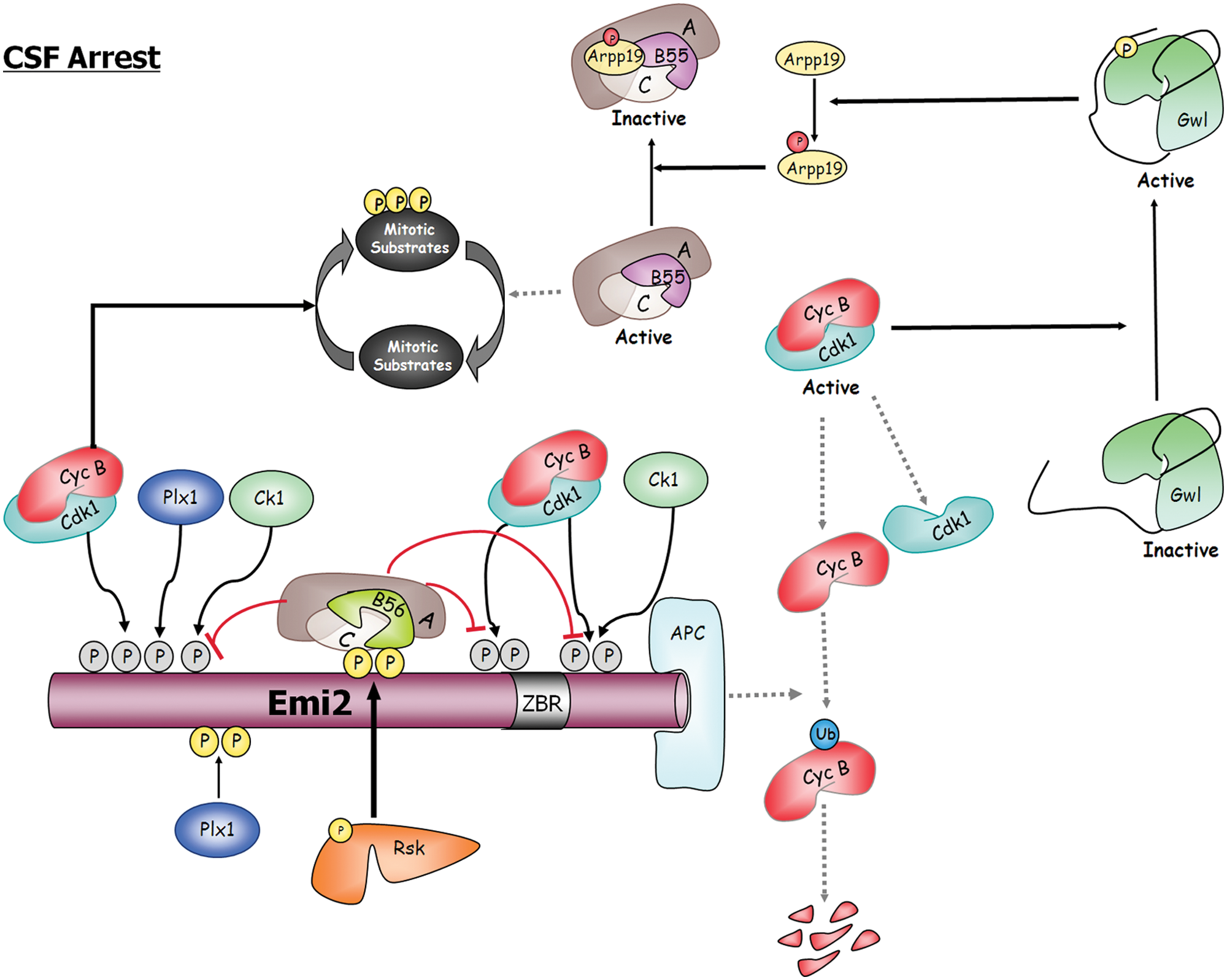
CSF arrest is the result of a tight balance between PP2A-B55 and PP2A-B56. Emi2 binds and inhibits the APC, the ubiquitin ligase of cyclin B. The inhibitory activity of Emi2 depends on the multiple phosphorylations of this protein by different kinases such as Plx1, cyclin B/Cdk1, CK1, and Rsk. Cyclin B-Cdk1, Plx1, and CK1 prevent Emi2 from binding the APC, whereas phosphorylation of Emi2 by Rsk stimulates the APC-inhibitory activity of this protein by recruiting PP2A-B56, the phosphatase responsible for the dephosphorylation of the cyclin B-Cdk1, Plx1, and Ck1 sites. In this way Rsk maintains cyclin B-Cdk1 activity and prevents reactivation of PP2A-B55, the phosphatase involved in substrate dephosphorylation at meiotic and mitotic exit. Dotted lines represent inactive pathways during CSF arrest. ZBR = zinc-binding region.
Oocyte fertilization induces SCFβ-Trcp-dependent degradation of Emi2, cyclin B proteolysis, and rapid inactivation of Gwl and Arpp19 by dephosphorylation. Reactivation of PP2A-B55 will then massively dephosphorylate cyclin B-Cdk1 substrates and promote meiotic exit.
Thus, metaphase II arrest is robustly maintained by a tight balance between the 2 phosphatases PP2A-B55 and PP2A-B56 that finely regulates protein phosphorylation.
Open Medical Therapies
Arpp19 and ENSA are intrinsically unstructured proteins (IUPs) that show little to no tertiary structure in isolation but assume function in concert with a conformational change induced by interaction with target molecules. IUPs can bind to multiple target molecules due to the plasticity of their structure. Induction of tertiary structure upon target binding can enhance specificity while keeping the binding affinity low, enabling interactions with multiple partners. 40
ENSA and Arpp19 bind the phosphatase PP2A.18,19 It is not known whether this binding takes places with the complex or with one of the subunits. Moreover, the mechanism by which this association inhibits the enzyme is completely unknown, although it is likely that as reported above, the induction of a tertiary structure of Arpp19 and ENSA upon binding can modify phosphatase activity. PP2A has been reported to function as a tumor suppressor. 41 Decreased PP2A activity due to an increased expression of its inhibitor SET has been reported to promote malignant growth of chronic myelogenous leukemia cells. 42 Similarly, overexpression of Arpp19 and ENSA could be involved in the tumoral process. Along this line, decreased PP2A activity could be restored by blocking Arpp19 and ENSA binding to PP2A.
In addition to having a potential role in the tumorigenesis process, ENSA appears to be involved in different neuronal pathologies. Along this line, ENSA protein is greatly decreased in frontal cortex and cerebellum of Alzheimer disease and in Down syndrome. 43 ENSA binds alpha-synuclein, a chaperone-like protein present in presynaptic neurons. 44 It is known that alpha-synuclein regulates neuronal synapsis by interacting with a variety of proteins such as tyrosine hydroxylase or PP2A phosphatase.45,46 Mutations of alpha-synuclein are associated with Parkinson disease, and this protein is the major fibrillar component of the Lewy bodies in Parkinson and Alzheimer diseases and Down syndrome. 47 Alpha-synuclein is also an IUP. Binding of ENSA to alpha-synuclein in the presence of phospholipids promotes the restructuring of the N-terminal part of alpha-synuclein into an amphipathic helix, suggesting a putative role of this interaction to confer to alpha-synuclein its normal neuronal function and to prevent alpha-synuclein aggregation, neurotoxicity, and Lewy body formation in vivo. 44
Further work on the mechanisms of interaction of ENSA and Arpp19 with their targets is fundamental to understand the structural changes involved in the modulation of their activities under normal physiological conditions. These data will be essential also for the development of new medical therapies to treat diseases such as cancer, Alzheimer disease, and Down syndrome. In this regard, the construction of small peptides targeting the interphase between Arpp19 and ENSA and PP2A or alpha-synuclein to reestablish their normal physiological activities would be of great therapeutic interest.
The inhibition of the Gwl kinase to prevent phosphorylation and activation of ENSA and Arpp19 proteins offers another open approach to restore PP2A activity under pathological conditions. However, this approach requires the development of high-throughput screenings directed to the identification of new drugs that specifically inhibit the Gwl kinase without affecting the other AGC kinases.
Conclusions
Only 1 year after the first data were reported about a role of PP2A-B55 and Gwl in mitotic division, the basic nodule of the pathway regulating this phosphatase activity was characterized. A major challenge now is to identify the mechanisms by which Arpp19 and ENSA binding to PP2A-B55 results in the inhibition of the phosphatase. It will be also important to determine how this pathway is regulated and whether other phosphatases besides PP2A-B55 are involved. Another emerging theme will be the investigation of other physiological processes in which this pathway could be implicated. Finally, identification by cell-permeable inhibitors and/or small interphase peptides would certainly provide a key step toward dissecting the function of this pathway and may also have significant therapeutic potential.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research in the laboratory of TL and AC has received funding from the Agence Nationale de la Recherche (Programme Blanc) and from The Ligue Nationale Contre le Cancer (Equipe Labellisée).