
Abstract
Hyperactivity of the Myc oncogenic transcription factor dramatically reprograms gene expression to facilitate cellular proliferation and tumorigenesis. To elicit these effects, Myc coordinates the activation and repression of an extensive network of protein-coding genes and, as has recently been appreciated, noncoding RNAs including microRNAs (miRNAs). Consistent with their ability to potently influence cancer phenotypes, the regulation of miRNAs by Myc affects virtually all aspects of the Myc oncogenic program, including proliferation, survival, metabolism, angiogenesis, and metastasis. This review will summarize the current understanding of the mechanisms underlying Myc-dependent transcriptional and posttranscriptional control of miRNAs and the resultant effects on tumorigenesis. As miRNAs are integral nodes in the transcriptional network controlled by Myc, modulating their activity represents a promising new approach for cancer therapy.
The c-MYC oncogene (hereafter referred to as MYC) encodes an evolutionarily conserved basic helix-loop-helix leucine zipper transcription factor that is commonly dysregulated in cancer, resulting in pleiotropic effects on cancer cell growth, proliferation, survival, angiogenesis, and metastasis. 1,2 Although Myc with its binding partner Max is known to regulate transcription of hundreds if not thousands of protein coding genes, small noncoding RNAs known as microRNAs (miRNAs) are now appreciated to be additional important components of the Myc target gene network. Recent work has demonstrated that Myc dramatically reprograms miRNA expression, resulting in significant augmentation of its protumorigenic activities. A single miRNA can destabilize and/or inhibit the translation of multiple messenger RNAs, simultaneously providing coordinated regulation of numerous genes. 3 Regulation of miRNAs therefore enhances the functional complexity of Myc’s role in tumorigenesis but also offers new therapeutic opportunities for the treatment of cancer.
miRNA Biogenesis and Function
Most miRNA-encoding genes structurally resemble protein coding genes. The primary miRNA transcripts (pri-miRNAs) are usually transcribed by RNA polymerase II (although a small number are produced by RNA polymerase III); they are generally several thousand nucleotides in length, and they are capped, polyadenylated, and frequently spliced. 4-8 Indeed, many protein-coding genes contain miRNAs within introns. miRNA processing is initiated in the nucleus with cleavage of pri-miRNAs by the microprocessor complex which includes as core components Drosha, an RNase-III endonuclease, and DGCR8, an RNA-binding protein. This cleavage releases precursor stem loop structures, known as pre-miRNAs, which are ~70 nucleotides in length. 9-11 Pre-miRNAs are then exported to the cytoplasm by exportin 5 in a RanGTP-dependent manner. 12,13 Within the cytoplasm, pre-miRNAs are further processed by the RNase-III enzyme Dicer, which removes the stem loop and produces a ~22 nucleotide RNA duplex. 10 One strand of the duplex, referred to as the guide strand, stably associates with an Argonaute (Ago) protein, while the opposite strand (referred to as the passenger strand or miRNA*) is discarded. 14-16 The resulting miRNA-Ago complex associates with target mRNAs through miRNA:mRNA base-pairing interactions, most often within 3′ UTRs. miRNAs bind to their targets with imperfect complementarity, although interaction between the 5′ end of the miRNA (especially nucleotides 2-7, known as the seed sequence) is usually required for efficient target regulation. Following recruitment of the miRNA-Ago complex, the target mRNA undergoes accelerated turnover and/or translational repression. Many excellent reviews providing further details on miRNA biogenesis and function can be found elsewhere. 4,17,18
Myc-Mediated Reprogramming of miRNA Expression
Prior to 2005, it was not clear that miRNAs could act as important downstream effectors of key signaling pathways that influence cancer phenotypes. Indeed, the principal mechanisms through which miRNA expression is regulated, including the identities of mammalian transcription factors that control miRNAs, were unknown. Nevertheless, emerging evidence pointed to an important role for miRNAs in cancer. Early on, miRNAs that regulate cellular differentiation, proliferation, and apoptosis had been discovered in C. elegans and Drosophila. 19,20 These processes are central to the pathophysiology of cancer, hinting at a role for miRNAs in human tumorigenesis. Indeed, specific miRNAs were later found to be deleted or amplified in tumor samples, 21-23 and reports of frequent miRNA dysregulation in specific cancer subtypes emerged. 24-26 Based on these findings, it seemed likely that miRNAs would be functionally integrated into the oncogenic programs of important cancer genes such as MYC.
The first demonstration that miRNAs could indeed function as critical components of oncogenic pathways came with the discovery that Myc directly activates transcription of the polycistronic miR-17-92 cluster. 27 Induction of these miRNAs occurs through the canonical mechanism whereby Myc binds directly to an E-box within the first intron of the gene encoding the miR-17-92 primary transcript. This locus encodes 6 mature miRNAs: miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1. 27,28 The miR-17-92 cluster has been duplicated during vertebrate evolution, resulting in 2 other paralogs, the miR-106a-363 and miR-106b-25 clusters. Mice deficient in miR-17-92 have defective B-cell development and die shortly after birth due to lung hypoplasia and ventricular septal defects. 29 In contrast, deletion of the miR-106a-363 or miR-106b-25 clusters does not result in any overt phenotype. Both miR-17-92 and miR-106b-25 are broadly expressed and have some functional redundancy since their combined deletion results in embryonic lethality. Expression of the miR-106a-363 cluster is undetectable in most tissues, and its physiologic functions are unknown.
Significant evidence now exists which convincingly implicates the miR-17-92 cluster as a bona fide oncogene. This cluster is frequently amplified and/or overexpressed in B-cell lymphomas and several solid tumors including breast, colon, lung, pancreas, prostate, and stomach. 23,30 Enforced expression of miR-17-92 in mice using transgenic or retroviral strategies results in lympho- proliferative disease and potently accelerates disease progression in the Eµ-myc B-cell lymphoma mouse model. 28,31 miR-17-92 expression also promotes tumorigenesis in solid tumor models. 32,33 As discussed in detail below, the inhibition of key targets following Myc-dependent activation of miR-17-92 augments tumorigenicity by promoting cell proliferation, survival, angiogenesis, and metabolic reprogramming. These data document an important role for the miR-17-92 cluster within the Myc target gene network and illustrate the highly significant contribution of miRNA control to the phenotypic output of a pathway that is critical for normal development and cancer.
Despite the importance of activation of the miR-17-92 cluster, this represents only one aspect of a much broader Myc-regulated miRNA network. Further studies have demonstrated that Myc activity results in repression of numerous miRNAs, including many with documented tumor suppressor activity including let-7 family members, miR-15a/16-1, miR-26a, miR-29 family members, and miR-34a. 34 Each of these miRNAs have been demonstrated to exhibit antiproliferative, proapoptotic, and/or antitumorigenic activity in a variety of settings. 35-39 Accordingly, rescuing expression of several of these miRNAs in Myc-transformed B lymphoma cell lines dramatically inhibits tumorigenesis. 34 As expected, given their diverse targets, these repressed miRNAs broadly impact Myc-mediated phenotypes, as will be highlighted in greater detail below.
miRNA expression has been reported to be globally reduced in some tumor samples and cell lines, 40,41 and experimental inhibition of the miRNA biogenesis pathway accelerates tumorigenesis in vitro and in vivo. 42 While genetic loss of function of miRNA processing components is one cause of impaired miRNA maturation in tumors, 43,44 it is likely that Myc hyperactivity also contributes to widespread downregulation of miRNA expression. The mechanisms through which Myc represses miRNAs are therefore of particular interest, since reversing these effects could have important therapeutic implications. Although further investigation is necessary, initial studies have indicated that Myc utilizes both transcriptional and posttranscriptional mechanisms to repress miRNA expression. 34,45 For example, several primary transcripts encoding Myc repressed miRNAs are similarly downregulated in the high Myc state, consistent with a transcriptional mechanism of repression. Accordingly, Myc associates directly with evolutionarily conserved promoter regions upstream of several repressed miRNAs, as revealed by primary transcript mapping and chromatin immunoprecipitation. 34 The most comprehensively understood mechanism of Myc-mediated transcriptional repression involves Miz1, a transcription factor which normally functions to activate transcription. Myc interacts directly with Miz1, displacing its coactivator p300 and thereby antagonizing Miz1 transactivation. Further recruitment of DNA methyltransferase 3A (DNMT3a) enforces transcriptional repression by inducing DNA methylation. 2 Future studies are necessary to determine whether interactions with Miz1 underlie Myc-mediated transcriptional repression of miRNAs.
Myc is also able to block the maturation of specific miRNAs without affecting transcription of the pri-miRNAs. Chang et al. observed that Myc activity resulted in repression of mature let-7 miRNAs even though expression of let-7 primary transcripts was unchanged. 34,45 Interestingly, the RNA binding proteins Lin28 and Lin28b were known to act as negative regulators of let-7 maturation, highlighting their candidacy as intermediaries in Myc-mediated repression of this miRNA family. Lin28/Lin28b interacts directly with let-7 pre-miRNA stem-loops and may regulate let-7 at multiple levels including Drosha and Dicer processing. 46-49 In addition, Lin28/Lin28b interaction recruits the 3′ terminal uridylyl transferase TUT4 to pre-let-7, resulting in uridylation and subsequent decay of the pre-miRNA. 50-52 Both Lin28 and Lin28b are direct Myc target genes and are required for Myc-mediated repression of let-7. 45,53 Repression of let-7 family members by the Myc-Lin28/Lin28b pathway promotes cellular proliferation, invasion, and metastasis.
Given the ability of individual miRNAs to regulate hundreds of targets, it is not surprising that the ability of Myc to broadly reprogram miRNA expression has dramatic and pleiotropic effects on cellular phenotypes. Although a full description of the known functions of all Myc-regulated miRNAs is beyond the scope of a single review, illustrative examples of how miRNA regulation has been integrated into the Myc oncogenic program will be provided in the ensuing sections (summarized in Figure 1 and Table 1).
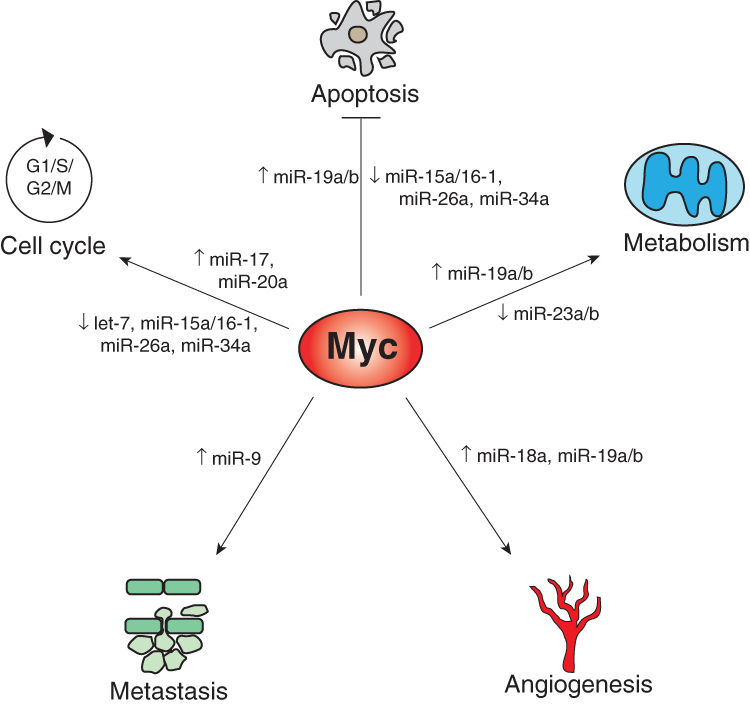
Myc-mediated induction or repression of miRNAs impinges upon multiple phenotypes that contribute to tumorigenesis, including cell-cycle progression, apoptosis, metabolic reprogramming, angiogenesis, and metastasis.
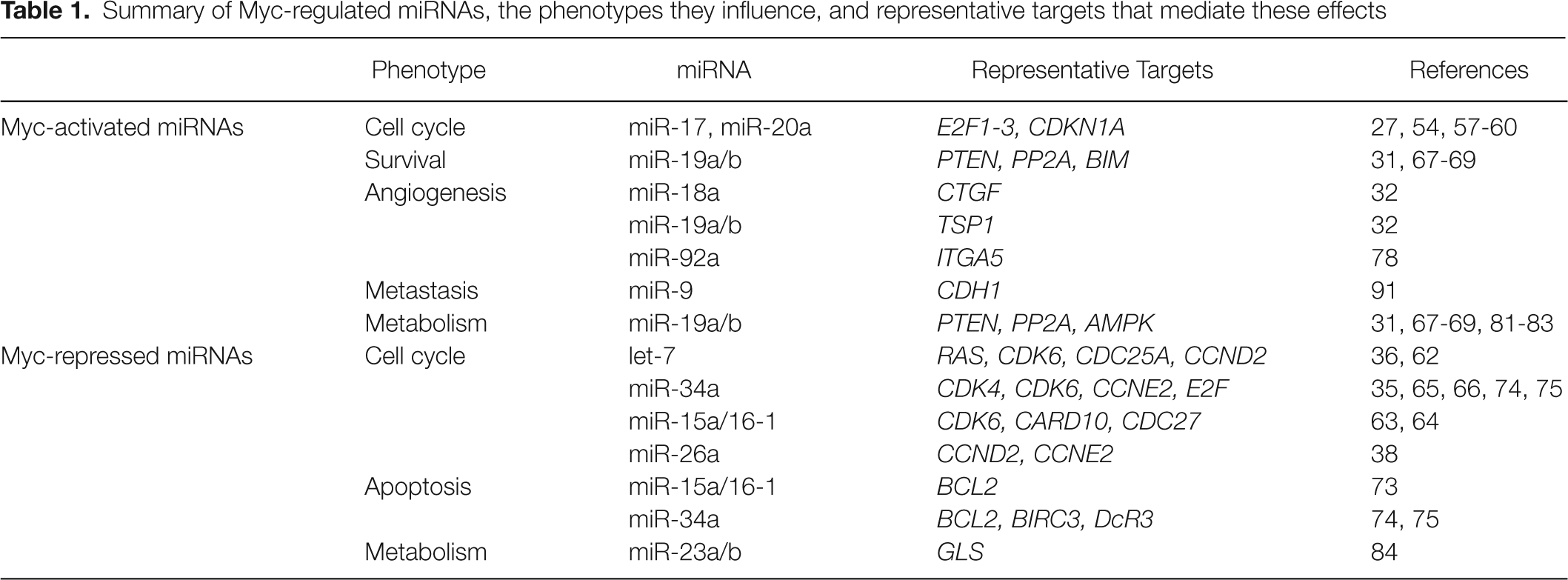
Summary of Myc-regulated miRNAs, the phenotypes they influence, and representative targets that mediate these effects
Cell-Cycle Control by Myc-Regulated miRNAs
Increased cellular proliferation is a major consequence of Myc activation in many cell types. Both induction and repression of specific miRNAs by Myc facilitates transit through the cell cycle through multiple mechanisms. For example, the initial studies which showed that miR-17-92 is directly induced by Myc also demonstrated that 2 components of the cluster, miR-17 and miR-20a, target the transcription factor E2F1. 27 Subsequent work also identified the related E2F2 and E2F3 transcription factors as additional targets of miR-17/20a. 54 E2F1-3, known as the activating E2Fs, positively regulate the transcription of genes required for DNA replication and are essential for G1-to-S phase progression. 55,56 However, their timing and levels of expression must be precisely controlled or, paradoxically, they can induce cell-cycle arrest or apoptosis. Interestingly, E2F1 is also known to be directly induced by Myc. Therefore, by simultaneously inducing E2F1 and its negative regulators, miR-17/20a, Myc may produce tightly controlled levels of E2F activity such that cell-cycle progression is promoted and apoptosis is avoided. Experimental support for this hypothesis was recently provided by Pickering et al., who showed that inhibition of miR-17 or miR-20a in serum-stimulated primary human fibroblasts results in a G1 arrest. 57 In this model system, inhibition of these miRNAs causes premature induction of E2F1, leading to accumulation of DNA damage and activation of the G1/S checkpoint.
miR-17 and related family members (miR-20a, miR-93, and miR-106b) also promote cell-cycle progression by targeting the cyclin-dependent kinase inhibitor p21 (CDKN1A). 58,59 p21 plays an essential role in p53-mediated cell-cycle arrest at both the G1/S and G2/M checkpoints. Therefore, through their ability to repress p21 levels, expression of these miRNAs overrides DNA damage induced cell-cycle arrest in multiple cell lines. 60 Conversely, loss of function of these miRNAs can induce activation of the G1 checkpoint. TGF-β signaling also leads to p21-dependent cell-cycle arrest in some contexts. Accordingly, overexpression or inhibition of miR-17 family members in gastric cancer cells inhibits or enhances TGF-β–dependent cell-cycle arrest, respectively. 59 These findings reveal a mechanism whereby activation of the miR-17-92 cluster by Myc impairs the ability of multiple tumor suppressor signaling pathways to restrain cancer cell proliferation.
Multiple miRNAs that are repressed by Myc, including let-7 family members, miR-15a/16-1, miR-26a, and miR-34a, have potent cell-cycle inhibitory activity. 38,61 Collectively, these miRNAs influence virtually all levels of the cell-cycle regulatory machinery. For example, key cell-cycle control factors regulated by let-7 include CDC25A, CDK6, CCND2 (cyclin D2), and many others. 62 miR-15a/16-1 inhibit expression of cell-cycle regulators such as CDK6, CARD10, CDC27. 35,63 miR-34a regulates CDK4, CDK6, CCNE2 (cyclin E2), and E2Fs, 64-66 while miR-26a represses CCND2 and CCNE2. 38 Thus, by downregulating expression of these miRNAs, Myc removes a major barrier to cell-cycle progression.
Myc-Regulated miRNAs in Apoptosis
In the Eµ-myc B lymphoma mouse model, ectopic expression of miR-17-92 strongly inhibits apoptosis in tumor cells without significantly affecting their proliferation. 28 Further studies have revealed that within the cluster, miR-19a and miR-19b-1 predominantly mediate the prosurvival activity in this model. 67,68 The antiapoptotic activity of miR-19 family members appears to be due in large part to the ability of these miRNAs to potentiate phosphatidylinositol-3-OH kinase (PI(3)K) signaling. Possibly the most important miR-19 target within this pathway is PTEN, which directly antagonizes PI(3)K activity by dephosphorylating the PI(3)K product phosphatidylinositol (3,4,5)-trisphosphate. 31,67-69 PTEN functions as a haploinsufficient tumor suppressor, underscoring the importance of maintenance of proper PTEN dosage to avoid PI(3)K pathway hyperactivity. Overexpression of miR-19b in Eµ-myc lymphoma cells leads to downregulation of PTEN and consequent induction of PI(3)K signaling, 68 whereas deletion of the miR-17-92 cluster in these cells leads to apoptosis, which is suppressed by short-hairpin mediated knockdown of PTEN. 67 Thus, miR-19-mediated repression of PTEN results in a potent survival signal in Myc-driven B-cell lymphoma.
A major effector of the PI(3)K pathway is AKT, which has many downstream prosurvival activities, including inhibition of the proapoptotic protein Bim. 70 The ϵ isoform of protein phosphatase 2A (PPP2R5E), which dephosphorylates and inactivates AKT as well as BIM, are targeted by miR-19a/b and by other miRNAs within the miR-17-92 cluster. 29,31,69,71 Similar to PTEN, Eµ-myc lymphoma cells are very sensitive to BIM dosage. Loss of a single allele of BIM is sufficient to suppress apoptosis and significantly accelerate disease progression in this model. 72 The miR-17-92 cluster is therefore able to reduce the dosage of key targets which operate at multiple levels in the PI(3)K pathway to increase the activity of this signaling cascade, thereby suppressing apoptosis.
Repression of miRNAs by Myc also contributes to cellular survival. For example, miR-15a/16-1, miR-34a, and miR-26a, which are repressed by Myc, 34 can each activate apoptosis in specific settings. The miR-15a/16-1 cluster directly downregulates the antiapoptotic protein Bcl-2, and expression of these miRNAs results in apoptosis in leukemic cell lines. 73 miR-34a is a direct transcriptional target of p53 and contributes to p53-dependent apoptosis. 74,75 In a mouse model of liver cancer, systemic delivery of miR-26a induced apoptosis specifically in tumor cells without affecting normal hepatocytes. 38 These observations highlight the broad impact of Myc-mediated miRNA reprogramming on cellular survival pathways.
Control of Angiogenesis by Myc-Regulated miRNAs
Myc promotes angiogenesis and vasculogenesis by upregulating the expression of proangiogenic factors such as vascular epidermal growth factor (VEGF) and angiopoietin 1/2, and by repressing antiangiogenic factors such as thrombospondin-1 (Tsp1) and connective tissue growth factor (CTGF). 32,76 Consistent with these activities, Myc-deficient mouse embryos exhibit vascular defects and die by embryonic day 10.5. 76 The miR-17-92 cluster plays an important role in Myc-induced angiogenesis. Dews et al. demonstrated that Myc potently stimulates angiogenesis and expression of miR-17-92 in a mouse orthotopic xenograft model of colon cancer. 32 miR-18a and miR-19 family members directly target the transcripts that encode CTGF and Tsp1, respectively, thereby reducing expression of these antiangiogenic proteins and allowing angiogenesis to proceed. Accordingly, enforced expression of miR-17-92 can partially replace the proangiogenic activity of Myc in this model. These findings were corroborated and extended in a study by Suárez et al., who showed that deletion of Dicer in mouse endothelial cells impairs physiologic and tumor-associated angiogenesis. 77 These effects are likely at least partially attributable to loss of the miR-17-92 cluster. VEGF signaling was demonstrated to upregulate miR-17-92, and expression of these miRNAs rescues cellular proliferation and capillary-like structure formation in endothelial cells lacking Dicer in vitro. The ability of miR-18a to regulate Tsp1 was also confirmed in this study. 77
In contrast to the proangiogenic activities of miR-18a and miR-19a/b, recent work has uncovered an antiangiogenic role for miR-92a, another miR-17-92 component. 78 Systemic inhibition of miR-92a in mice using antagomiRs (modified antisense miRNA inhibitors 79 ) stimulates angiogenesis following multiple insults, including limb ischemia and myocardial infarction. The antiangiogenic activity of miR-92a is partially attributable to its ability to downregulate the angiogenesis promoting factor integrin α5. Precisely how the activities of the proangiogenic (miR-18a, miR-19a/b) and antiangiogenic (miR-92a) components of the miR-17-92 cluster are coordinated will be an important issue for future study. It is possible that in different cellular contexts, specific members of the cluster accumulate to different levels, resulting in distinct phenotypic outcomes. Additionally, differential expression of targets could certainly contribute to context-dependent effects of miR-17-92 expression on angiogenesis.
Metabolic Reprogramming by Myc-Regulated miRNAs
Glucose and glutamine are the 2 major carbon substrates utilized by highly proliferative cells. 80 As described in detail above, Myc-dependent activation of the miR-17-92 cluster potentiates signaling through the PI(3)K-AKT pathway. 31,67-69 miR-19 family members play a major role in this activity by virtue of their ability to downregulate the expression of PTEN, which antagonizes PI(3)K activity. PI(3)K-AKT signaling strongly promotes glucose metabolism and fatty acid synthesis through multiple mechanisms, including increasing glucose transporter surface expression and enhancing glycolytic enzyme activity. 81 AKT also activates ATP citrate lyase, an enzyme that is required for glucose dependent fatty acid synthesis and tumor growth in vivo. 82
Glutamine metabolism is promoted by Myc through several mechanisms, including the direct transcriptional activation of glutamine transporters. 83 Myc activity also leads to increased expression of mitochondrial glutaminase (GLS), a key enzyme that converts glutamine to glutamate, which serves as a substrate in the TCA cycle for the production of ATP. 83,84 Both glutamine and GLS are required for Myc-mediated cancer cell survival and proliferation. Interestingly, Myc increases GLS activity by directly repressing transcription of 2 miRNAs that target the GLS transcript, miR-23a and miR-23b. 84 Therefore, Myc coordinates both glucose and glutamine metabolism through miRNA regulation to promote bioenergetics necessary for cell proliferation and survival.
Regulation of miRNAs by N-Myc
The MYC homolog MYCN (encoding the N-Myc oncoprotein) is frequently amplified in neuroblastoma, a tumor type which accounts for 15% of all pediatric cancer deaths. 85 MYCN is potently oncogenic in this setting, and its amplification is predictive of poor prognosis. N-Myc activity is associated with specific miRNA expression signatures, 86,87 and N-Myc and c-Myc have been shown to share several common miRNA targets. For example, the miR-17-92 cluster is directly induced by N-Myc and exhibits oncogenic activity in this tumor type. 87-90 Both N-Myc and c-Myc also induce expression of miR-9. 91 Through its ability to target E-cadherin, activation of this miRNA promotes migration and invasiveness of breast cancer cells. miR-9–mediated repression of E-cadherin also stimulates β-catenin signaling, which induces VEGF expression and angiogenesis. Consistent with these observations, miR-9 levels correlate with MYCN amplification and metastatic disease in patients with neuroblastoma. Another miRNA induced by both N-Myc and c-Myc is miR-421. 87 miR-421 represses expression of the ataxia-telangiectasia mutated (ATM) kinase, which plays a critical role in the DNA damage response, 92 suggesting that expression of this miRNA might contribute to genomic instability. These findings indicate that, similar to their roles in the c-Myc pathway, miRNAs act as effectors of the N-Myc oncogenic program.
Targeting Myc-Driven Tumorigenesis with miRNA Therapeutics
The ability of miRNAs to potently influence cancer phenotypes suggests that strategies designed to inhibit or augment the activity of specific miRNAs in tumor cells might provide therapeutic benefit. Given the pleiotropic oncogenic functions of the miR-17-92 cluster and its importance in the Myc network, targeting these miRNAs with systemically delivered antisense reagents such as antagomiRs or locked nucleic acid oligonucleotides 79,93 represents a promising approach worthy of evaluation. Studies of therapeutic inhibition of miR-17-92 in tumor models in vivo have not yet been reported. More progress has been made in the development of so-called “miRNA replacement” therapies, which aim to restore the activity of specific miRNAs whose expression is lost in cancer cells. This strategy would be expected to complement small-molecule therapies, which inhibit oncogenic signaling, as miRNA replacement promotes the reestablishment of tumor suppression mechanisms. Moreover, since miRNAs regulate hundreds of targets in multiple pathways, the emergence of resistant tumor cell clones might be unlikely, as many simultaneous mutations would be necessary to overcome the effects of miRNA expression. miRNA replacement is especially attractive in the context of Myc-mediated transformation because it has been demonstrated that Myc activity results in widespread repression of miRNA expression, and reexpression of individual miRNAs in Myc-transformed cells inhibits tumorigenesis in xenograft assays. 34 miRNA replacement therapies have now been successfully used to treat cancer in multiple mouse models, highlighting the potential for translating this approach into patient therapies.
The ability of miRNA replacement therapies to suppress Myc-driven tumorigenesis in vivo is strongly supported by studies by Kota et al., 38 who implemented this approach in a Myc-transgenic liver cancer model. 94 This work focused on miR-26a, a miRNA previously demonstrated to be repressed by Myc in lymphoma models. 34 miR-26a is downregulated in human and mouse liver tumors, and low expression of this miRNA is associated with poor prognosis in patients with hepatocellular carcinoma. 38,95 Expression of this miRNA in human liver cancer cells in vitro induces a G1 arrest associated with direct targeting of cyclins D2 and E2. Adeno-associated virus (AAV)–mediated delivery of this miRNA to liver tumors in the mouse results in dramatic suppression of disease progression. While miR-26a administration activated apoptosis in tumor cells, normal hepatocytes did not experience any obvious toxicity. Although further work is necessary to elucidate the molecular basis for this specificity, it is likely that the high level of natural miR-26a in hepatocytes confers tolerance to additional expression of this miRNA. In contrast, loss of miR-26a in tumor cells is clearly necessary to maintain their malignant potential. Given the documented safety of AAV vectors (clinical trials using this delivery platform have been performed and are underway) 96,97 as well as the apparent safety of miR-26a–based therapy in the mouse, clinical testing of AAV-mediated miR-26a delivery for the treatment of hepatocellular carcinoma is clearly warranted.
The effectiveness of miRNA replacement therapies is not limited to Myc-driven tumorigenesis. Delivery of let-7 has been shown by multiple laboratories to be an effective therapeutic approach in a well-established mouse model of non–small-cell lung cancer driven by oncogenic Kras mutations. Intratracheal delivery of let-7 using lentiviral or adenoviral vectors strongly suppresses tumor initiation and maintenance in this model. 98-100 These effects appear to be due primarily to the ability of let-7 expression to inhibit cancer cell proliferation rather than by inducing apoptosis. 100 Given the lack of effective therapies for this tumor type and the ability to locally deliver agents to the lung via inhalation, delivery of let-7 through this route represents a highly promising treatment for this tumor type.
As described in this review and elsewhere, the expression of many distinct miRNAs can inhibit cancer cell proliferation and survival in a large number of tumor types, underscoring the broad potential of miRNA replacement therapies. Although the lack of efficient nucleic acid delivery strategies for many tissue sites currently represents a major obstacle, the development of improved delivery methods is a major area of investigation in academic and pharmaceutical laboratories, offering hope for future implementation of this approach for many types of cancer.
Conclusion
Myc-mediated reprogramming of miRNA expression introduces a newly appreciated layer of complexity to the diverse physiologic and pathologic functions of this oncoprotein. The network of Myc-regulated miRNAs control key effectors of this oncogenic pathway that influence the cell cycle, apoptosis, metabolism, angiogenesis, and metastasis. Further study of the precise functions of these miRNAs will be essential to achieve a complete understanding of Myc-driven tumorigenesis and will likely reveal new diagnostic and therapeutic opportunities for cancer.
Footnotes
J.T.M. is an HHMI Early Career Scientist and a Rita Allen Foundation Scholar. The Mendell laboratory receives additional support from the NIH (R01CA120185 and P01CA143292).
The authors declare no conflicts of interest with respect to the publication of this article.