
Abstract
MYC is a key regulator of cell growth, proliferation, metabolism, differentiation, and apoptosis. MYC deregulation contributes to breast cancer development and progression and is associated with poor outcomes. Multiple mechanisms are involved in MYC deregulation in breast cancer, including gene amplification, transcriptional regulation, and mRNA and protein stabilization, which correlate with loss of tumor suppressors and activation of oncogenic pathways. The heterogeneity in breast cancer is increasingly recognized. Breast cancer has been classified into 5 or more subtypes based on gene expression profiles, and each subtype has distinct biological features and clinical outcomes. Among these subtypes, basal-like tumor is associated with a poor prognosis and has a lack of therapeutic targets. MYC is overexpressed in the basal-like subtype and may serve as a target for this aggressive subtype of breast cancer. Tumor suppressor BRCA1 inhibits MYC’s transcriptional and transforming activity. Loss of BRCA1 with MYC overexpression leads to the development of breast cancer—especially, basal-like breast cancer. As a downstream effector of estrogen receptor and epidermal growth factor receptor family pathways, MYC may contribute to resistance to adjuvant therapy. Targeting MYC-regulated pathways in combination with inhibitors of other oncogenic pathways may provide a promising therapeutic strategy for breast cancer, the basal-like subtype in particular.
Introduction
According to SEER data projections for 2009, an estimated 192,370 women were diagnosed with breast cancer and 40,170 died of the disease. Breast cancer development is a complex process involving multistep genetic and epigenetic changes that drive normal breast cells into highly malignant derivatives. Based on gene expression profiles, breast cancer has been classified into 5 subgroups: luminal A, luminal B, Her2/Neu amplified, basal-like, and normal-like. 1 Each group has distinct biological features and clinical outcomes, suggesting that breast cancer progresses through different molecular pathways among patients. 1,2 To fulfill the goal of personalized therapies, a better understanding is warranted of the interacting pathways that contribute to aggressive behavior and poor outcomes in breast cancer.
c-MYC (MYC) was identified as v-MYC avian myelocytomatosis viral oncogene homolog almost 3 decades ago. 3 As the most influential transcription factor, MYC regulates up to 15% of human genes and is involved in cell growth, proliferation, metabolism, differentiation, and apoptosis. 4 MYC is a potent activator of tumorigenesis, and its deregulation has been found in a variety of cancers. 4–6 Numerous reports have revealed the critical role MYC plays in breast tumorigenesis and progression (reviewed in References 7–9). We briefly summarize the role of MYC in breast cancer, particularly as it relates to different molecular subgroups.
MYC Protein and Biological Function
MYC protein is a transcription factor with basic region/helix–loop–helix/ leucine zipper domain. The MYC protein activates transcription as part of a heteromeric complex with MYC-associated factor X (Max), which binds to E-boxes with the consensus core sequence CACGTG or with noncanonical sequences. 10 Activation involves the recruitment of multiple coactivators and protein complexes to E-box elements. 11,12 Although the role of MYC in the activation of transcription is clearly established, the mechanisms of transcriptional repression by MYC are not well understood. 13 MYC may repress gene expression through tethering with MYC-interacting zinc finger protein-1 (Miz-1) and inhibits the gene transactivation by Miz-1. 11
As illustrated in Figure 1, MYC plays multiple roles in breast cancer development and progression. MYC is a key regulator of cell proliferation and transformation. 4,6,11 Numerous genetic targets for MYC activation and repression have been identified, and its expression has been associated with activation of cell cycle regulators. 4 MYC controls G1-S transition through activating downstream targets such as cyclin E/Cdk2 and repressing cyclin-dependent kinase (Cdk) inhibitor p21Cip1. This appears to be the major mechanism governing MYC’s effects on cell cycle progression in breast cancer cells. 14,15 MYC also facilitates cell proliferation through repression of transcription of the p27Kip1 cdk inhibitor gene. 16 In addition, MYC promotes cell cycle progression by activation of cyclin D1, Cdk4, Cdc25A, E2F1, and E2F2. 6 Yet, D-type cyclins/Cdk4 are downstream effectors of MYC in transformation. MYC failed to transform fibroblasts lacking cyclin Ds, 17 and lack of Cdk4 inhibits MYC tumorigenic activities in epithelial tissues. 18 MYC expression in human mammary epithelial cells induces an epithelial-to-mesenchymal transition (EMT) accompanied with dramatic change in cell morphology. E-cadherin expression is repressed by MYC through a posttranscriptional mechanism, and E-cadherin repression is necessary for MYC-induced cell transformation. 19 p21Cip1 attenuates Ras- and MYC-dependent breast tumor EMT and breast cancer stem cell–like gene expression in cultured cells and transgenic mice. 20
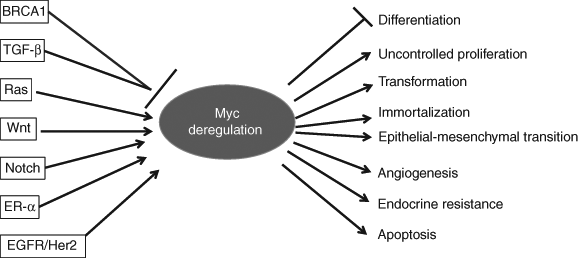
MYC regulatory network in breast cancer. MYC is regulated in multiple levels in breast cancer cells. TGF-β and BRCA1—which can repress MYC gene expression and MYC’s transcriptional activity, respectively—are frequently inactivated by mutation or epigenetic mechanism in breast cancer. Yet, oncogenic pathways such as Ras, Wnt, Notch, ER-α, and EGFR/Her2 positively influence MYC expression or stability. Loss of tumor suppressors and activation of oncogenic pathways can cause MYC deregulation. Deregulated MYC promotes tumor growth, metastasis, and therapy resistance. BRCA1 = breast cancer susceptibility gene 1; TGF-β = transforming growth factor β; Ras = rat sarcoma; Wnt = wingless-type murine mammary tumor virus integration site family; ER-α = estrogen receptor α; EGFR = epidermal growth factor receptor; Her2 = human epidermal growth factor receptor 2.
In addition, MYC blocks cell differentiation through repressing differentiation-induced p21Cip1 expression via Miz-1-dependent interaction with the p21Cip1 core promoter. 21 MYC is also involved in cell immortalization. Telomerase, which preserves chromosome integrity by maintaining telomere length, is a direct target of MYC. MYC activates telomerase by inducing expression of its catalytic subunit, telomerase reverse transcriptase (TERT). TERT and MYC can induce immortalization when constitutively expressed in transfected cells. 22 In breast tumors, expressing high levels of TERT mRNA is correlated with increased levels of MYC mRNA. 23 Furthermore, MYC promotes oncogenesis through regulating cell growth, metabolism, and metastasis. 4,11 MYC is essential for angiogenesis, and vascular endothelial growth factor (VEGF) expression levels are controlled by MYC. 24 A study revealed that MYC plays a central role in the angiogenic switch in tumors. 25 MYC activates miR-9, a negative regulator of the key metastasis suppressor E-cadherin, and leads to increased cell motility, invasiveness, as well as enhanced angiogenesis. The miR-9 miRNA can induce an elevated expression of VEGFA through activating β-catenin signaling. 26
However, MYC induces apoptosis in the absence of survival factors. 27–29 Both mitochondrial apoptotic pathway and death receptor–mediated apoptosis pathway are involved in MYC-mediated apoptosis. MYC can modulate the intrinsic apoptotic pathway by altering the balance of pro- and antiapoptotic members of the Bcl-2 family, in parallel with or independent of p53. MYC promotes death receptor–mediated apoptosis by repressing survival pathways activated by death receptors. 30 Abrogation of MYC-induced apoptosis is crucial for cellular transformation and tumorigenesis.
MYC Deregulation in Breast Cancer
Gene Amplification
MYC is located on chromosome 8q24, which is frequently translocated or amplified in a variety of cancers. 31 Translocation involving MYC locus has not been reported in breast cancer thus far. However, MYC gene amplification and overexpression in breast cancer were first reported in 1986. 32 Since then, a large body of literature has emerged regarding gene amplification and correlation with clinicopathological features. Unfortunately, the frequency of MYC amplification and its prognostic relevance in human breast cancer have been inconsistent. In an effort to clarify the clinical significance of MYC amplification in breast cancer, Deming et al. conducted a meta-analysis of 29 studies. The weighted average frequency of MYC amplification in breast tumors, which was defined as a 2-fold increase in gene copy number, was 15.7% (95% CI = 12.5%–18.8%). 33 MYC amplification exhibited significant but weak associations with tumor grade (RR = 1.61), lymph node metastasis (RR = 1.24), negative progesterone receptor status (RR = 1.27), and postmenopausal status (RR = 0.82). Amplification was significantly associated with risk of relapse and death, with pooled estimates of RR = 2.05 (95% CI = 1.51–2.78) and RR = 1.74 (95% CI = 1.27–2.39), respectively. 33 Further studies have confirmed the correlation between MYC amplification and poor prognosis. 34–37 In particular, MYC amplification appears to be a strong prognostic marker of early recurrence in node-negative breast cancer patients. 35 In contrast, other reports showed that there was no association of MYC amplification with established risk factors. 38,39 MYC amplification was not significantly associated with overall survival of patients with invasive cancer. 40,41 Thus, it is important to define amplification threshold in studies evaluating MYC amplification in breast cancer.
MYC amplification appears to play a role in breast cancer progression because it has been observed in the more aggressive phenotype of ductal carcinoma in situ 42 or in only the invasive component. 34,37,43 We have shown that MYC amplification is a frequent event in breast tumors from BRCA1 germ-line mutation carriers and in sporadic tumors with BRCA1 inactivation owing to BRCA1 promoter hypermethylation. 44,45 We observed MYC amplification in 53% of BRCA1-mutated tumors, compared with 23% in sporadic tumors. Of the sporadic cases with MYC amplification, 57% were BRCA1 promoter methylated. Overall, MYC amplification was found in a significantly higher proportion of tumors with BRCA1 dysfunction (48% versus 14%, P = 0.0003). Thus, it is plausible that MYC amplification occurs after loss of BRCA1 during the progression of tumorigenesis.
Transcriptional Regulation and mRNA Stabilization
MYC gene amplification, as detected by fluorescent in situ hybridization (FISH), is significantly associated with overexpression of its mRNA and protein. 46 MYC gene amplification has been found in about 15% of tumors, whereas 22% to 35% of tumors have reported overexpression at the mRNA level. 47–49 Protein overexpression has been reported in about 40% of breast tumors, 38,50 which indicates that mechanisms other than gene amplification, such as transcriptional regulation and mRNA and protein stabilization, are involved in MYC overexpression.
The regulation of the MYC promoter is complex yet poorly understood. Many signaling pathways, transcription factors, and cis regulatory elements are involved in regulating MYC transcription. 12 The MYC promoter is a target of the Wnt signaling pathway, Notch pathway, Ras/Raf/MAPK, NF-κB pathway, and TGF-β (Figure 1). These pathways are all critical in breast cancer and may be important druggable targets.
Downregulation of MYC is a critical event for growth inhibition induced by TGF-β and is frequently impaired in cancer cells. TGF-β represses the MYC promoter through the Smad3/Smad4/E2F-4,5/DP-1/p107 complex and consequently induces cell cycle arrest. 51,52 The Smad complex that mediates MYC repression is a target of oncogenic signals in breast cancer because formation of this complex is deficient in transformed breast cells. TGF-β-dependent MYC repression is lost in breast cancer cell lines. 53 In addition, decreased nuclear Smad 3 is correlated with high tumor grade, larger tumor size, and hormone receptor negativity. 54 Ras antagonizes TGF-β-dependent repression of MYC transcription through interfering with the TGF-β at multiple levels. 55,56
In addition, constitutive Wnt/β-catenin signaling is one of the most frequent abnormalities in human cancers. 57 MYC has been shown to be a downstream effector of β-catenin in colorectal cancer. 58–61 MYC deletion rescues adenomatous polyposis coli (APC) deficiency in the small intestine. 62 The cross talk between MYC and β-catenin has been extensively investigated in colorectal cancers and hepatocellular carcinomas that show constitute activation of β-catenin owing to mutations of Wnt pathway components. 57,63 Even though mutations of Wnt components such as APC, β-catenin, and Axin are rare in breast cancer, an elevated level of nuclear and/or cytoplasmic β-catenin was detected by immunohistochemistry (IHC) staining in breast carcinomas and correlated with poor patient outcome. 64,65 An autocrine Wnt signaling necessary for oncogenic transformation was identified in breast cancer cell lines. 66,67 Another study showed that MYC activates Wnt in breast cancer by suppressing the Wnt inhibitors DKK1 and SFRP1, which are strongly repressed in breast cancer cell lines. MYC-dependent repression of DKK1 and SFRP1 is accompanied by Wnt target gene activation and endogenous T-cell factor activity. Derepression of DKK1 or SFRP1 reduces MYC-dependent transforming activity. 68 Based on these observations, a positive feedback loop of MYC and Wnt signaling was proposed in breast cancer. 69 Yet, stable expression of SFRP1 in MDA-MB-157 and MDA-MB-231 breast tumor cell lines blocks proliferation of both cell lines and results in reduced MYC mRNA in MDA-MB-157 and decreased MYC protein in MDA-MB-231. 66,70 However, how the canonical Wnt pathway, which is dependent on β-catenin stabilization, regulates MYC in breast cancer and its biological significance are still unclear.
In colorectal cancer cells, MYC mRNA is stabilized by coding region determinant-binding protein (CRD-BP), which is overexpressed in colon cancer and has been identified as a new target of β-catenin/TCF signaling. 71 Gene amplification of CRD-BP has been reported in breast cancer 72,73 ; however, association between CRD-BP expression and MYC mRNA stabilization is under investigation.
Notch signaling plays an essential role in regulating cell fate, proliferation, apoptosis, and migration. 74 The pathway is activated through the interaction of Notch receptors with Delta-like and Jagged ligands on neighboring cells. 75 The Notch intracellular domain (NICD) is released by proteolytic cleavages and enters into the nucleus. In the nucleus, NICD forms a transcriptional activator complex with RBP-Jκ/CBF1 and Mastermind and activates transcription. 76 The role for Notch signaling in human breast cancer has been suggested by the development of adenocarcinomas in the murine mammary gland following pathway activation 77 and the loss of Numb expression, a negative regulator of the Notch pathway, in a large proportion of breast carcinomas. 78 In addition, accumulation of NICD in a variety of human breast cancer cell lines and tissue samples suggests that the pathway is aberrantly activated in human breast cancer. Interestingly, blocking Notch signaling pathway by overexpressing Numb reverts the tumorigenic phenotype of MCF7 and MDA-MB-231 breast cancer cell lines. 79 In one study, high-level coexpression of Jagged1 and Notch1 was observed in human breast cancer and was associated with poor overall survival. 80
Upregulation of MYC was observed in NICD-induced tumors, and conditional ablation of MYC in the mammary epithelium prevented the induction of regressing NICD neoplasms, suggesting that MYC is a downstream effector in Notch-induced mammary tumorigenesis. Following molecular analyses revealed that mouse and human MYC genes are direct transcriptional targets of NICD acting through its downstream CBF1 transcriptional effector. Consistent with this observation, Notch1 and MYC expression was shown to be positively correlated in 38% of examined human breast carcinomas 77 ; however, the immunostaining data did not correlate with any clinicopathological features of the tumors. The interaction between Notch and MYC in breast cancer development and progression deserves further study.
Protein Overexpression and Stabilization
Protein overexpression has been found in 45% of breast tumors with IHC 50 and in 42% with Western blot. 38 Because of technical difficulties and breast tumor heterogeneity, the correlations between MYC protein levels and clinicopathological characteristics are less consistent than those between such characteristics and MYC amplification. As reviewed by Efstratiadis et al. in 2007, 9 the interpretation of IHC results obtained with mouse monoclonal antibody 9E10 was problematic, and consistent results were achieved through a rabbit polyclonal antibody (N262, Santa Cruz Biotechnology, Santa Cruz, CA). With N262 antibody, we have observed specific nuclear staining of primary breast tumors and cultured breast cancer cells by IHC and immunofluorescence staining (Xu et al., in preparation).
MYC protein is subject to different posttranslational modifications, including phosphorylation, ubiquitinylation, and acetylation. Those modifications affect not only its stability but also its functions, such as interaction with other proteins and transcriptional activity. 81 In addition to upregulating MYC at the mRNA level through antagonizing TGF-β as discussed above, Ras enhances the accumulation of MYC activity by stabilizing the MYC protein. 82 MYC is stabilized with phosphorylation at Ser62 by MAPK, a Ras downstream effector. Subsequent phosphorylation at Thr58 by glycogen synthase kinase 3β (GSK-3β) destabilizes MYC. 83 Ras inhibits GSK-3β through PI-3K pathway and blocks MYC protein degradation. 11 A complex signaling cascade involving GSK-3β, the Pin1 prolyl isomerase, and the PP2A-B56α phosphatase controls the degradation process. 84 It has been recently shown that tumor suppressor scaffold protein Axin1 plays a crucial role in facilitating the formation of the degradation complex. 85 The ubiquitin E3 ligase Fbw7 ubiquitylates phopho-58 MYC and targets it for proteasomal degradation. 86 The loss of Fbw7 in cancer cells leads to MYC activation and promotes cell proliferation during tumorigenesis. 87 Consistent with this, high-level MYC expression was detected from SUM149PT, an Fbw7-mutant breast cancer line, and forced expression of Fbw7 reduced the levels of MYC. 88 Stabilization of MYC in primary breast tumors has not been evaluated. High-quality commercial anti-phospho Ser62 MYC antibody needs to be developed to facilitate such studies.
Role of MYC in Breast Cancer Subtypes
MYC overexpression has been suggested through DNA microarray analysis to correlate with distinct molecular breast cancer subtypes. Studies have shown MYC to be highly expressed in basal-like tumors (7 of 14, 50%) and normal-like cancers (5 of 13, 38%) yet low in luminal (5 of 47, 11%) and Her2-overexpressing (1 of 11, 9%) types. 89,90 In collaboration with Chuck Perou, we have analyzed the gene expression pattern with a large cohort of primary breast tumors (N = 168) by DNA microarray and confirmed that MYC is highly expressed in the basal-like subtype in comparison to the other 4 breast cancer subtypes (Figure 2; Xu et al., in preparation), which suggests that MYC is involved in subtype-specific pathways.
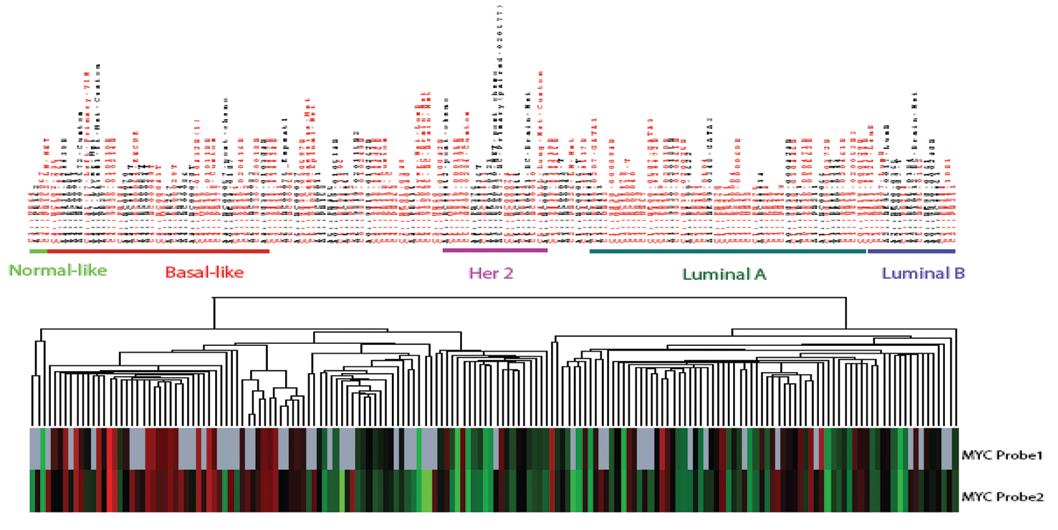
MYC mRNA is highly expressed in basal-like tumors. RNAs were extracted from 168 primary breast tumors and applied for microarray analysis (Agilent, Santa Clara, CA). Gene expression data were retrieved from the database after the Lowess normalization procedure and data filtering. Upregulated and downregulated genes were picked up by the significant analysis of microarray 2-classes unpaired t test. Unsupervised hierarchical clustering demonstrates segregation of MYC expression in discrete groups. Red = Caucasian; Black = African American.
MYC, BRCA1, and Basal-Like Tumors
Basal-like breast cancer is characterized by the expression of genes normally found in breast basal myoepithelial cells. 1,89 At the protein expression level, with IHC analysis as a surrogate, basal-like breast cancer is defined by tumors that are triple negative (ER-α negative, progesterone receptor negative, and Her2/Neu negative), cytokeratin 5/6 positive, and/or epidermal growth factor receptor (EGFR) positive. 91 Basal-like breast cancer constitutes one of the most challenging subtypes of breast cancers. Although it accounts for only 15% of breast cancer cases, basal-like breast cancer is responsible for a disproportionate number of breast cancer deaths. 92 This subtype of tumors typically has an early-age onset, 93,94 a strong tendency to metastasize to other organs, such as the brain and lung, 95,96 and a lack of current therapeutic targets. 92,97,98 Thus, prognosis of basal-like breast cancer is poorer than that of other breast cancer subtypes, 99,100 and there are currently no effective therapeutic strategies for it. 101 Combination chemotherapy remains the only therapeutic option.
With analysis of multiple gene expression data sets, it has been shown that activation targets of MYC are more frequently overexpressed in poorly differentiated tumors than in well-differentiated tumors. In breast cancers, this ES-like signature is associated with high-grade estrogen receptor–negative tumors—particularly, the basal-like subtype. 102 A meta-analysis showed elevated activity of MYC and E2F in the basal-like breast cancer subgroup, 103 which suggests that MYC may play an important role in the development of basal-like breast cancer.
Interestingly, aberrant β-catenin expression has been found in basal-like tumors. 65 Currently, we are addressing how MYC is regulated by canonical Wnt pathway in basal-like cancer cells. Elevated mitotic index and geographic tumor necrosis are morphologic features associated with the basal-like subtype. Those phenomena may be linked to MYC overexpression, which drives uncontrolled proliferation or apoptosis. With an overexpression model, it has been reported that Wnt signaling cooperates with MYC in oncogenic transformation by inhibiting MYC-induced apoptosis. 104 This process is accompanied by upregulation of β-catenin and other downstream targets, such as Cox-2 and WISP. Because β-catenin and MYC are overexpressed in basal-like cancer cells, it would be intriguing to investigate whether endogenous β-catenin can suppress MYC-induced apoptosis.
BRCA1 is involved in multiple cellular processes, including DNA repair, cell cycle control, apoptosis, and transcriptional regulation. 105–109 Loss of BRCA1 can result in genomic instability and contribute to breast cancer initiation and progression. 106,110,111 Women with BRCA1 mutations have a 50% to 80% chance of developing breast cancer in their lifetime. 112,113 Inherited mutations of the BRCA1 gene account for about 5% of all breast cancer cases. 114 In addition, BRCA1 is downregulated by promoter methylation in 11% to 31% of sporadic breast cancers. 115,116 Reduced BRCA1 expression correlates with disease progression. 117 We have demonstrated that inactivation of BRCA1 by promoter hypermethylation contributes to a subset of sporadic breast cancers, with a resulting molecular and clinicopathologic phenotype similar to that of hereditary BRCA1-associated breast cancers. 45 Expression microarray analyses have indicated strong similarities in gene expression between BRCA1-mutated cancers and sporadic basal-like cancers. 2 The majority of breast cancers arising in BRCA1 germline mutation carriers have clinicopathological features similar to those of basal-like cancers, including triple negativity, EGFR overexpression, high-grade tumors, and poor prognosis. 93,118,119 The similarity between BRCA1-mutated and basal-like cancers indicates that disruption of BRCA1 may be a common initial pathogenic event. 118,119 It is still unclear how normal cells are transformed, given that deregulated expression of MYC alone is not enough to convert normal cells into tumor cells. 120 Mouse mammary tumor virus–MYC-driven tumor has a long latency, suggesting that MYC alone is not sufficient to drive tumor development. 121 Additional genetic alternations, such as loss of tumor suppressor BRCA1, may be required for tumor formation.
BRCA1 binds to MYC and inhibits its transcriptional and transforming activity. 122 A complex of BRCA1, Nmi, and MYC is required to inhibit MYC-induced human TERT gene promoter activity in breast cancer. 123 In addition, BRCA1 and MYC act together to transcriptionally repress psoriasin, which is related to chemotherapeutic agent sensitivity. 124 These observations demonstrate the crucial role of BRCA1 as a tumor suppressor in preventing MYC-induced breast cell transformation. As previously discussed, we have observed MYC amplification in BRCA1-mutated and promoter methylated tumors. 44 MYC overexpression and BRCA1 loss seem highly correlated in a large portion in basal-like breast cancers. It is tempting to hypothesize that loss of BRCA1 with MYC overexpression leads to development of breast cancer—especially, basal-like breast cancer.
Because MYC and BRCA1 are 2 proteins that significantly contribute to tumorigenesis, the functional consequence of MYC–BRCA1 interaction during breast tumor progression is of great interest. How MYC regulates BRCA1 is unclear, however. Interestingly, among the numerous target genes identified by a serial analysis of gene expression screen, BRCA1 is one of the MYC-activated gene targets. 125 Because MYC transcription activity could be repressed by BRCA1 through protein interaction, it is most likely that BRCA1 could serve as a feedback repressor on a MYC-activated gene such as TERT. Presumably, in normal mammary epithelial cells, the activation of TERT expression by MYC would be suppressed by BRCA1, which can be activated by MYC. TERT overexpression could be led by disruption of this repression owing to inherited BRCA1 mutations or BRCA1 loss in sporadic breast cancer cases mainly caused by BRCA1 promoter methylation. Enhanced TERT catalytic activity could be one of the early steps contributing to cell immortalization.
MYC, EGFR, and Her2/Neu
EGFR family of receptor tyrosine kinases are associated with cell proliferation, cell survival, angiogenesis, and cell motility. 126 The family members include EGFR (Her1), Her2, Her3, and Her4, which form homodimers or heterodimers upon ligand-dependent activation. Interestingly, EGFR and Her2 link to distinct subtypes of breast cancer with poor prognosis.
EGFR protein overexpression has been reported to occur in 16% to 36% of breast cancers. 127 As discussed above, EGFR overexpression is a feature of basal-like tumors. 91 Consistent with this is the report that 65% to 72% of basal-like tumors have EGFR overexpression. 128 In our laboratory, EGFR expression was found in 35% of 129 unselected breast cancer cases from the University of Chicago Medical Center. Strikingly, 84% of basal-like tumors showed EGFR overexpression. 129 The correlation between EGFR overexpression and prognosis is still uncertain in breast cancer. However, a recent study showed that overexpression of EGFR is associated with poor prognosis in basal-like tumors. 129
EGFR regulates MYC by activating the Ras/Raf/MEK/ERK pathway and phosphatidylinositol 3 kinase–Akt (PI3K-Akt) pathway. ERK activation drives cell proliferation through transactivation of the cyclin D1 gene and MYC. 130 It has been found that activated Akt provides protection from MYC-mediated apoptosis in association with upregulation of the Bcl-xL protein in MYC-overexpressing murine mammary epithelial cells. Blockade of EGFR kinase activity with pharmacological inhibitors results in a significant induction of cellular apoptosis. 131 It has recently been reported that EGFR inhibitor gefitinib strongly induces apoptosis and represses telomerase activity in MDA-MB-231 cells via transcriptional downregulation of TERT through MYC and via posttranslational modification of TERT protein through inactivation of Akt. 132 Because both EGFR and MYC are overexpressed in basal-like breast cancer, it is important to study their correlation—especially, the potential antagonism in regulating apoptosis.
HER2 overexpression, mainly owing to amplification, has been found in 20% to 30% of breast cancers. 133 Her-2/neu amplification and MYC amplification are characteristic features of nonlobular breast cancers. 134 MYC amplification has been significantly associated with Her2 amplification and closely linked with cell proliferative activity, as measured by the Ki67 labeling index. The strong correlation between MYC and Her2 amplification and proliferative activity indicates a biological link between these genes in breast cancer cells. 41 It appears that Her2/MYC-coamplified cancers have a worse prognosis than that of tumors with only one of these amplifications, but the data are inconsistent, probably because of the nonstandardization of assays for MYC amplification. 36
HER2 subtype of breast cancer is characterized by high-grade tumors, resistance to endocrine therapy, and worse prognosis. 133,135–137 Patients with HER2-positive tumors have greatly benefited from the development of trastuzumab (Herceptin), a humanized monoclonal antibody directed against Her2. 138 Herceptin has proven to increase survival among women with metastatic breast cancer and to significantly increase the disease-free survival of patients with Her2-positive early breast cancer. 135–137 Herceptin induces G1 cell cycle arrest of Her2 overexpression breast cancer cells through induction of p27Kip1 and reduction of Cdk2. 139,140 Further study showed that trastuzumab inhibits cyclin D and MYC expression through the PI3K and MAPK pathways, releases the sequestrated p27Kip1 protein from cyclin D-Cdk4/6 complexes, and increases the effect of p27Kip1 on Cdk2-cyclin E complexes. 141 Multiple mechanisms are involved in trastuzumab resistance, including P27kip1 downregulation in breast cancer cells. 142–144 Amplifications of MYC, EGFR, HER2, CCND1, and TOP2-A have been identified from a trastuzumab-resistant breast cancer cell line (B585) by comparative genomic hybridization and FISH analysis. 145 However, whether MYC amplification or overexpression correlates with trastuzumab resistance needs further investigation.
MYC and ER-α
ER-α and its ligand estrogen play critical roles in breast cancer pathogenesis, progression, and treatment. Hormonal therapy via estrogen depletion or selective estrogen receptor modulators is widely used to block the action of estrogen in women with hormone responsive breast cancers. 146 MYC is a downstream effector of ER-α, and MYC promoter contains an atypical estrogen-response cis-acting element. 147 MYC is upregulated by estrogen in ER-α-positive breast cancer cells, and it plays a critical role in estrogen-induced breast cell proliferation. 148,149 Antisense oligonucleotides directed against MYC inhibit estrogen-induced cell proliferation in a manner similar to that of antiestrogens. 150,151 MYC protein is stabilized in response to estrogen and phospholipase D in MCF-7 breast cancer cells. Estrogen and phospholipase D suppress phosphorylation of MYC at Thr58 and prevent MYC from proteasome degradation. 152 Gene expression analysis of estrogen-treated or MYC-induced breast cancer cells showed that half the estrogen-regulated genes were regulated by MYC, indicating that a significant component of estrogen-induced mitogenesis is mediated by MYC. 153 A meta-analysis of gene expression from 5 large microarray data sets relative to ER-α status revealed that increased transcriptional activity of MYC is a characteristic of basal-like breast cancers; namely, it mimics a large part of an estrogen response in the absence of ER-α, suggesting a mechanism by which these cancers achieve estrogen independence. 103
Endocrine resistance has been a major problem for hormonal therapy. Many factors are involved in the process, including loss of ER-α expression by epigenetic regulation, ligand-independent activation of ER-α, and upregulation of growth factor signaling pathways. 154 Cross talk between the ER-α and the EGFR/HER2 signaling pathways contributes to the development of resistance to endocrine therapies. The acquired resistance is significantly inhibited by treatment with the EGFR/HER2 inhibitor gefitinib, lapatinib, or the monoclonal anti-Her2 antibody trastuzumab. 155,156 The amplification of growth factor receptor signaling cascades can converge on activation of MYC, thus potentially influencing endocrine responsiveness. Overexpression of the estrogen-targeted cell cycle regulatory molecules MYC and cyclin D1 has been associated with altered sensitivity to endocrine therapy, and overexpression can modulate sensitivity to clinically relevant antiestrogens in in vitro models. 154 Inducible expression of either MYC or cyclin D1 is sufficient for S-phase entry in cells previously arrested in G1 phase by pretreatment with an estrogen antagonist in MCF-7 cells. MYC or cyclin D1 activates cyclin E-Cdk2 by promoting the formation of high-molecular-weight complexes without the Cdk inhibitor p21Cip1. 157 Another study showed that MYC alone is sufficient to confer antiestrogen resistance in human breast cancer. Induced MYC expression stimulates cell growth in the presence of estrogen antagonist ICI 182,780. 158 Mechanistically, aberrant MYC expression can contribute to antiestrogen resistance by repressing p21Cip1 expression. Expression of p21Cip1 blocks MYC-mediated cell cycle progression in the presence of antiestrogen. In antiestrogen-treated cells, the elevated mRNA and protein levels of p21Cip1 decrease upon either MYC induction or estrogen treatment. MYC is required for estrogen-mediated decreases in p21Cip1. Because aberrant MYC expression is frequently observed in human breast cancers, it can contribute to antiestrogen resistance by altering p21Cip1 expression. 15 Furthermore, sustained expression of MYC has been observed in the development of tamoxifen-resistant cell lines, suggesting that these molecular changes may contribute to clinical antiestrogen resistance. 159 Recently, a cooperative involvement of ER-α and MYC in mediating estrogen regulation of VEGF expression and function has been found in breast cancer cells and tumors. Long-term overexpression of MYC alone can partially mimic estrogen regulation of VEGF, suggesting an additional mechanism for antiestrogen resistance. 160 However, all data of antiestrogen resistance are from ectopic overexpression of MYC in the breast cancer cell line MCF-7, and correlation between MYC expression and endocrine resistance in clinical specimens remains unclear. 154
Therapeutic Strategy
As summarized above, MYC is deregulated in breast cancer through multiple mechanisms and likely contributes to breast cancer development and prognosis. However, MYC is essential for normal cell function. Furthermore, like a double-edged sword, deregulated MYC can promote cell proliferation or cause apoptosis depending on different circumstances. This complicates targeting MYC for therapeutic purposes, and additional caution needs to be paid. Nevertheless, therapeutic interventions that target MYC expression and function have been extensively studied—such as inhibiting mRNA transcription, destabilizing MYC protein, and interfering with protein–protein interaction of MYC-Max. 161
Antisense oligonucleotides (ASOs), triple helix–forming oligonucleotides (TFOs), and small interfering RNA (siRNA) have been tested in breast cancer cells. Treating MCF-7 breast cancer cells with an ASO repressed MYC expression and led to decreased proliferation, suggesting that ASOs have potential clinical application. 151 A plasmid-based siRNA against MYC reduced its expression in MCF-7 cells by up to 80%. The rate of colony formation in soft agar and that of tumor growth in nude mice were significantly reduced. Importantly, depletion of MYC by siRNA promoted apoptosis of MCF-7 cells upon serum withdrawal, which indicates that targeting MYC with siRNA has therapeutic potential in the treatment of human breast cancer. 162 ASOs and siRNA both have high specificity; however, several issues need to be resolved before they can be applied in a clinical setting: Compared with siRNA, ASOs confer a risk of immunogenicity. siRNA is unstable in plasma and hard to deliver, which could be improved by fusing multiple siRNAs targeting MYC, Mdm2, and VEGF mRNA to a positively charged protein covalently linked to a specific antibody. 163
TFOs bind to double-stand purine-rich promoter regions and block transcription factor binding. TFOs are unstable, and covalent linkage of a TFO to daunomycin could increase stability of the triple helix and activity of the oligonucleotide in cells. The daunomycin-conjugated TFO inhibits transcription in vitro and reduces MYC promoter activity in MCF-7 and MDA-MB-231 breast cancer cells. 164 Interestingly, TFO significantly enhances effectiveness of the anticancer nucleoside analogue gemcitabine. SKBR3 cells treated in combination with TFOs, and gemcitabine showed a reduction in cell survival and capacity for anchorage-independent growth. 165 Decoy oligodeoxynucleotides (DOs) compete for the binding to DNA and attenuate the effect of overexpressed transcription factor in tumor cells. Double-strand DOs with the MYC consensus site reduce the proliferation of MCF-7 cells in a dose-dependent manner. 166 However, all the above-cited studies were conducted with non-basal-like cancer cells, which have lower MYC expression compared with that of basal-like cancer cells. In our view, targeting MYC in basal-like tumor may have a profound effect.
Aside from inhibiting MYC directly, targeting other oncogenic pathways that cross talk with MYC has great potential for therapeutic strategy. Inhibition of aberrant Wnt pathway activity in cancer cell lines efficiently blocks cell growth and induces apoptosis. Drugs that disrupt the β-catenin–TCF pathway have been extensively tested for the treatment of colorectal cancers. 167 Sulindac is the most extensively studied nonsteroidal anti-inflammatory drug in the context of chemoprevention. The efficacy of sulindac as an anticancer agent can be attributed to its inhibition of cancer cell proliferation and induction of apoptosis. 168 Another β-catenin inhibitor, ICG-001, efficiently inhibits growth of colon carcinoma cells in vitro, in the Min mouse and nude mouse xenograft models of colon cancer, 169 which makes it an attractive lead compound for the development of new cancer chemotherapeutics. If it turns out that β-catenin could suppress MYC-induced apoptosis, targeting β-catenin with small molecular inhibitors would be an effective therapy against basal-like tumors. EGFR also inhibits MYC-mediated apoptosis through upregulation of the Bcl-xL protein in MYC-overexpressing murine mammary epithelial cells. 131 Inhibiting EGFR kinase activity with pharmacological inhibitors, such as gefitinib and lapatinib, or with monoclonal antibody cetuximab may induce apoptosis in a subset of basal-like cancer cells that have both MYC and EGFR overexpression. The drawback is that inhibition of the EGFR pathway may cause downregulation of MYC. Because Notch1 and MYC expression is positively correlated in human breast cancers, 77 inhibition of Notch signaling may be a therapeutic strategy. Interestingly, a recent study showed that the Wnt pathway inhibitor ICG-001 could antagonize NICD signaling. 170
Compared with cells of other breast cancer subtypes, basal-like breast cancer cells have an activated Ras-like transcriptional program and show greater sensitivity to a selective inhibitor of MAPK/ERK (MEK). However, treatment with a selective MEK inhibitor causes upregulation of PI3K pathway signaling in PTEN-mutated basal-like cancer cells. Blockade of both PI3K and MEK signaling potently impairs the growth of basal-like breast cancer cells in vitro and in vivo. 171 Because the Ras/Raf/MAPK pathway positively regulates MYC on multiple levels, correlation between MYC status and drug sensitivity is of interest.
Conclusion
MYC deregulation contributes to breast cancer initiation and progression and is associated with poor outcomes. Because of the complexity of MYC regulation and the heterogeneity of breast cancer, many questions remain unanswered. For example, why is MYC disproportionally overexpressed in basal-like breast tumors? What drives MYC overexpression? How can we target it for therapeutic purpose? Understanding the biology of basal-like breast cancer—especially, the mechanism of MYC deregulation in basal-like tumors—will lead us to find suitable therapeutic targets and develop effective treatment strategies against this aggressive form of breast cancer. In addition, MYC may be involved in endocrine resistance and trastuzumab resistance, which are major challenges for adjuvant therapy in breast cancer treatment. Further mechanism studies should lead to the design of more effective MYC-targeting strategies, which have the potential to reduce the breast cancer morbidity and mortality.
Footnotes
Acknowledgements
We apologize to colleagues whose work was not cited because of limited space. We thank Michelle Porcellino for critical reading of the article.
The Olopade laboratory is supported by the National Cancer Institute’s Specialized Program of Research Excellence in Breast Cancer, the Breast Cancer Research Foundation, the Entertainment Industry Foundation’s National Women’s Cancer Research Alliance, and the Falk Medical Research Trust.
The authors declared no potential conflicts of interest with respect to the authorship and publication of this article.