
Abstract
Tumors have been recently recognized as aberrant organs composed of a complex mixture of highly interactive cells that in addition to the cancer cell include stroma (fibroblasts, adipocytes, and myofibroblasts), inflammatory (innate and adaptive immune cells), and vascular cells (endothelial and mural cells). While initially cancer cells co-opt tissue-resident vessels, the tumor eventually recruits its own vascular supply. The process of tumor neovascularization proceeds through the combined output of inductive signals from the entire cellular constituency of the tumor. During the last two decades, the identification and mechanistic outcome of signaling pathways that mediate tumor angiogenesis have been elucidated. Interestingly, many of the genes and signaling pathways activated in tumor angiogenesis are identical to those operational during developmental vascular growth, but they lack feedback regulatory control and are highly affected by inflammatory cells and hypoxia. Consequently, tumor vessels are abnormal, fragile, and hyperpermeable. The lack of hierarchy and inconsistent investment of mural cells dampen the ability of the vessels to effectively perfuse the tumor, and the resulting hypoxia installs a vicious cycle that continuously perpetuates a state of vascular inefficiency. Pharmacological targeting of blood vessels, mainly through the VEGF signaling pathway, has proven effective in normalizing tumor vessels. This normalization improves perfusion and distribution of chemotherapeutic drugs with resulting tumor suppression and moderate increase in overall survival. However, resistance to antiangiogenic therapy occurs frequently and constitutes a critical barrier in the inhibition of tumor growth. A concrete understanding of the chief signaling pathways that stimulate vascular growth in tumors and their cross-talk will continue to be essential to further refine and effectively abort the angiogenic response in cancer.
Keywords
Introduction: Tumors Are Organs
The transformation of normal cells into a neoplasm and subsequently into a malignant tumor is a stepwise process through which the tumor acquires what Hanahan and Weinberg named the “hallmarks of cancer”. 1 In order for tumors to succeed in situ, they have to develop ways to sustain proliferative signaling, evade antiproliferative safeguards, resist apoptotic programming, achieve replicative immortality, and summon new vasculature to import nutrients and export waste. If a tumor is able to gain the necessary mutations to migrate and survive in new niches, it can then take advantage of available vascular networks and travel to colonize new frontiers (Fig. 1). In fact, death from cancer is due less to the primary tumor outcompeting surrounding tissue and more due to cachexia, immune suppression, and thromboembolisms.2,3
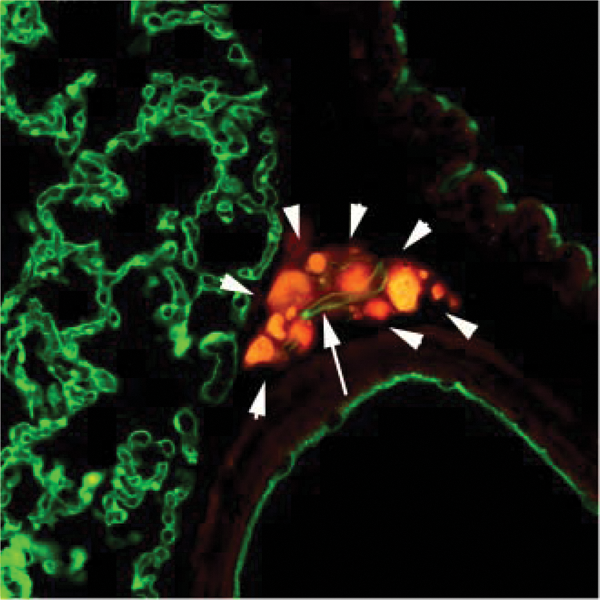
Tumor angiogenesis in the lung. Prostate epithelial cells are labeled in red, and vasculature is labeled with anti-PECAM antibodies in green. Note that a very small cluster of tumor cells (white arrowheads) has recruited a single vessel (arrow).
As interdisciplinary information reached cancer biology, the field has moved away from the concept of cancer as an isolated self-sufficient ball of aberrant cells. Currently, tumors are viewed as “organs” composed of multiple and highly interactive cell types.3-6 Thus, the tumor is made up of the primary cancer cells and of a court of stromal cells that actively contribute to its maintenance. The tumor stroma includes mesenchymal derived cells (like fibroblasts, adipocytes, and smooth muscle cells), inflammatory cells (innate and adaptive immune cells), and vascular cells (endothelial cells and pericytes). Each of these cell types can be found in normal stroma; but in a tumorigenic setting, the cancer has appropriated, modified, and corrupted these cells to do its bidding.
The cancer cell element of the tumor organ, through growth factor paracrine signaling, stimulates the differentiation of fibroblasts from a tumor-suppressing cell to a tumor-supporting cell. These cancer/tumor-associated fibroblasts (CAFs or TAFs) are characterized by smooth muscle α actin, fibroblast activation protein, and Thy-1 expression.7,8 The acquisition of these features is gradual and, at some point, irreversible. First, upon a breach in tissue integrity, tissue-resident fibroblasts become myofibroblasts due to the upregulation of smooth muscle α actin. Myofibroblasts are normally involved in wound healing and display an increased capacity for secreting extracellular matrix (ECM) proteins and cytokines, such as IL-6 and RANTES, which relay distress calls to the immune system. If healing is resolved, myofibroblasts revert back to tissue- resident fibroblasts.7,8 However, continued pressure from tumor cells can impose additional changes on myofibroblasts that now evolve into CAFs. These can no longer revert into tissue fibroblasts and, in addition, gain resistance to apoptosis. 8 The ECM secreted by CAFs can hoard and store cytokines and growth factors produced by the tumor and the assortment of stromal cells in the microenvironment. CAFs also secrete matrix metalloproteinases (MMPs) that further enable tumor invasion. 9 Degradation of the ECM also makes stored growth factors and cytokines biologically available to spark proliferation and migration via the activation of signal transduction cascades. 9 The presence of CAFs, along with the influx of inflammatory cells to the tumor site, inspired the concept of the tumor as “a wound that never heals”. 10
In addition to the cancer cells and fibroblast components, the tumor organ has a substantial inflammatory cell constituency. The notion that immune cells aid in cancer growth is counterintuitive if we think of the immune system as a mechanism to fight against foreign and abnormal insults. In fact, customarily, immune cells do identify and eliminate abnormally growing cells. 6 A tumor only develops once the cancer cells have evolved to bypass immune recognition and have co-opted inflammatory cells to their own advantage. 3 The inflammatory cells seen in the tumor microenvironment include tumor-associated macrophages (TAMs), mast cells, eosinophils, neutrophils, and dendritic cells.3,11 TAMs most closely resemble the M2 branch. 3 They secrete cytokines and growth factors that induce tumor growth, invasion, metastasis, and angiogenesis. They also have been shown to suppress cytotoxic T-cell activity.3,11 In addition to TAMs, N2 lineage neutrophils and mast cells release ECM-degrading enzymes and angiogenesis-stimulating factors.3,11 As a consequence, the recruitment of immune cells to the tumor amplifies an already upregulated network of growth factor cross-talk that promotes tumor growth.
Tumor-associated vascular beds provide the tumor with nutrients and oxygen and with a mode of waste removal. If a tumor develops in a well-vascularized region, the co-option of pre-existing vessels might be employed. Otherwise, angiogenesis, or the branching of new blood vessels from established vascular networks, is a necessity for tumor expansion beyond 400 µm. 12 The induction of vascular growth is only possible by building a proangiogenic environment through the collective effort of tumor cells, CAFs, and resident inflammatory cells. 7 Growth factors secreted by the tumor create a chemotactive gradient to recruit endothelial cells and pericytes away from their existent vascular beds. In addition, MMPs generate a large variety of matrix protein fragments that facilitate migration and vascular morphogenesis.3,7 Once a vascular network is established, it can further support the tumor by shuttling in nutrients and oxygen and exporting away waste products. With the aid of the cooperative tumor microenvironment, the cancer continues to have the opportunity to accumulate mutations and metastasize. The fact that angiogenesis is required for tumors to achieve two of the “hallmarks of cancer” highlights its importance in cancer progression. Understanding the tumor as an organ requires an understanding of angiogenesis. This review will summarize the current knowledge on the molecular mechanisms that underlie this process.
The Tumor Vasculature
Within a tumor, immune, stromal and cancer cells, immune and stromal cells engage in altruism, the goal of which is to nurture cancer cell growth and facilitate expansion of the tumor to alternative niches. 1 Blood vessels are an essential component of this goal, as they provide metabolic means and routes for metastatic expansion. Nonetheless, how frequent is “true” angiogenesis (growth of new blood vessels) in tumors? Is the formation of new blood vessels necessary for tumor growth? Or are local tissue-resident vessels sufficient to feed tumor cells?
While the notion that angiogenesis is triggered as soon as the tumor reaches 0.4 mm is conceptually appealing, it is unclear how frequent new blood vessels versus existent blood vessel utilization occurs in tumors (Fig. 2). New vascular invasion is essential for tumors originating from avascular tissues such as the epidermis. Yet, even in this environment, once tumor cells disrupt the basement membrane and invade the underlying dermis, a wealth of dermal resident vessels is available to the incipient tumor cells. Vascular co-option is prominent in carcinomas originating from single epithelia (e.g., many types of breast cancer) as well as in any tumor of mesenchymal origin (sarcomas). Thus, as tumor cells expand, they progressively take over the local vasculature through co-option of vessels 13 (Fig. 2). However, it should be stressed that these co-opted vessels are no longer normal. The association of tumor cells with resident vessels alters their morphology, physiology, and responses to therapy.14,15 It is critical to acknowledge that we do not know how much co-option versus tumor-initiated vascular growth occurs in any given tumor. It is experimentally difficult to distinguish between the 2 processes. However, a future clear delineation between co-option and new vascular sprouting might be particularly important for better selection of therapeutic intervention.
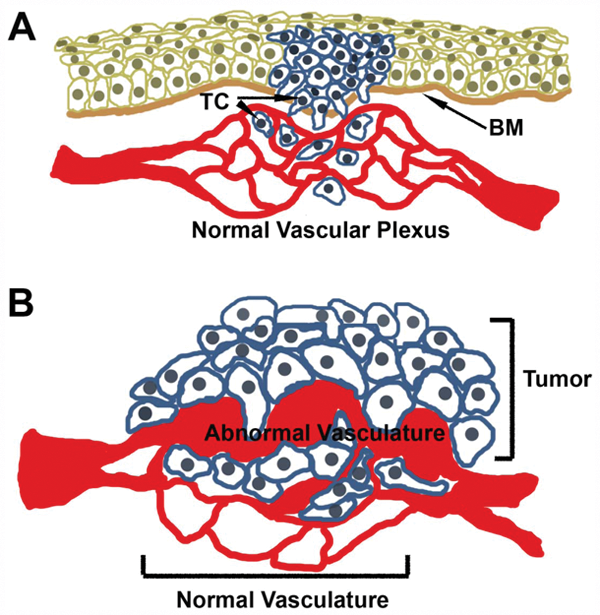
Co-option of normal vasculature by tumor cells. (
Whether co-opted or the result of legitimate angiogenesis, tumor-associated vessels differ from normal vessels morphologically and functionally. Normal vessels are hierarchic, evenly distributed, and because of their effective coating of smooth muscle cells/pericytes, are able to efficiently deliver nutrients to the tissue they serve. In contrast, tumor vasculature tends to be inefficient because of structural transformations induced by paracrine signaling and tumor-endothelium cell contact. Tumor vessels tend to be unevenly distributed and form chaotic, tortuous networks with irregular branching patterns. 16 Tumor vessels exhibit bidirectional blood flow and are not constantly perfused. 17 Using artificial methods like tumor cell injection or adenoviral delivery of VEGF into mice, investigators have been able to generate surrogate vessels to understand the pathology of blood vessels in crisis. Whereas adenovirally delivered VEGF (adeno-VEGF) mimics an acute spike of VEGF (the most prominent angiogenic growth factor), as seen in wound healing, injection of tumor cells is meant to model the chronic, sustained exposure of vessels to VEGF. Adeno-VEGF injection creates a fast and amplified production of VEGF that tapers off to normal levels after about a month. 16 These studies have shown “tumor” blood vessels to be larger than normal vessels, with an altered surface area to volume ratio that results in poor nutrient delivery and waste removal. 16 Efforts have been made to categorize and follow the evolution of these surrogate vessels as a method of understanding the vessels commonly found in tumors.
Using morphological and physiological criteria, investigators have identified 6 distinct types of tumor vessels16,18: 1) Mother vessels (MVs) are the first type of vessels to form after exposure to high levels of VEGF and/or after injection of tumor cells. These vessels have been also seen in healing wounds and human tumors. They are maintained as long as high concentrations of VEGF164 are present. MVs originate from pre-existing capillaries and venules and form in a 3-step process. After injection, there is an initial peak in cell proliferation after about 3 to 5 days. To accommodate a 3- to 5-fold increase in vessel diameter, the MV must shed the associated pericytes and degrade the vessel basement membrane (VBM). Cathepsin cystein proeases, secreted by pericytes, are responsible for this degradation of the VBM. Normally, cystein protease inhibitors (CPIs) would prevent VBM degradation, but they are found at low levels at the injection site. The individual endothelial cells lining the vessel wall have to stretch and flatten. To accommodate the increase in endothelial cell surface area, vesiculovacuolar organelles fuse with plasma membrane. MVs are transitional vessels that evolved into 1 of 3 types of daughter vessels. 2) After VEGF levels start to fall, capillaries are formed in a process called luminal bridging: when MV endothelial cells extend processes across the lumen. The blood flow gets divided into the capillaries, which can sustain in the presence of low VEGF levels. 3) Alternatively, in the presence of high levels of VEGF, MVs can evolve into glomeruloid microvascular proliferations. These vessels are formed when an overproliferation of CD31-VEGFR2+ endothelial cells bridge the lumen of MVs. These smaller vessels acquire pericytes and an excess of abnormal VBM. When VEGF levels fall, these vessels regress into capillaries. 4) Vascular malformations, the last type of daughter vessel, form when MVs obtain a thin asymmetrical coat of smooth muscle cells that might supply the endothelial cells with VEGF. Vascular Malformations (VMs) maintain the large diameter of MVs and can survive independent of an exogenous source of VEGF. 5) and 6) Feeder arteries and draining veins are vessels directly upstream and downstream from the tumor. They are large, tortuous vessels that are not a product of angiogenesis.
Functionally, tumor vessels are inappropriately permeable to large macromolecules and are inefficient at oxy- genating the tumor and clearing waste. Tumor-associated vessels leak not just water but “exudate” a fluid close to the composition of whole plasma. 19 This is in contrast to normal vessels that are able to filter large macromolecules and prevent them from passing into the tissue. In the tumor microenvironment, plasma proteins like fibrinogen leak into the stroma and encounter tissue factor, which polymerizes fibrinogen into fibrin gel clots, which aids in tumor expansion in a number of ways 19 : a) The fibrin gel absorbs water and prevents the clearance of fluid, causing edema and a pressure gradient that favors diffusion from the interstitial space into the vessel lumen. This is in direct contrast to the function of a normal blood vessel, which should deliver nutrients to the tissue. b) Fragment E, cleaved from fibrin by MMPs, is directly angiogenic. c) Growth factors can become trapped in the gel, protecting them from degradation and making them available to transform stromal cells. So, not only are tumor vessels unable to perform their normal functions, but they also actively contribute to the formation of the tumor-promoting microenvironment. Tumor vessels are unable to effectively oxygenate tumor tissues and are also inefficient at removing waste metabolites because of the aforementioned structural abnormalities. 20 Some areas of the tumor are chronically hypoxic and acidic because they are hypovascular, and other regions are acutely hypoxic and acidic due to intermittent blood flow. 20 One can imagine how this hostile microenvironment could select only for the most aggressive tumors that are able to switch to signaling pathways that allow adaptation. In addition, both hypoxia and acidosis induce the expression of growth factors like VEGF, Ang2, PDGF, PlGF, TGFα, IL8, and HGF in vitro. 21 These growth factors can induce both cancer cell and stromal cell proliferation. In mice, it has been shown that low pH controls VEGF promoter activity by way of the Ras-ERK1/2-AP1 pathway, while hypoxia induces VEGF promoter activity via the HIF1α route. 22 So, as the tumor shapes the microenvironment to promote abnormal blood vessels, the deviant vasculature in turn promotes a microenvironment that selects for and nurtures a more aggressive cancer phenotype.
The abnormal tumor vasculature is a formidable barrier to chemotherapeutic agents administered to fight the tumor. Hypoxia itself can cause resistance to radiation and other therapies. 23 Certain DNA-hydrolyzing chemotherapeutic agents can only work in the presence of oxygen, and the highly acidic microenvironment can attract weakly basic drugs, preventing access to the cancer cell targets. 20 The chaotic structure of the vasculature, poor smooth muscle cell/pericyte coverage, and the dysfunctional ability of the endothelium to transport fluid create a high intratumoral pressure that perpetuates difficulties in perfusion of the growing tumor.23-25 Due to the hyperpermeability of tumor vessels and the focal leaks often seen in tumor vessels, blood flow rates are reduced as measured by red blood cell velocity.20,26 Studies have shown that tumors often have increased interstitial pressure 23 and that they can affect the interstitial pressure of surrounding normal tissue. 25 Hydraulic conductivity studies using intratumoral injection of Evans blue dye into tumor centers have shown that tumors vary in their resistance to fluid flow. 24 This resistance could be relieved using hyaluronase to hydrolyze the ECM, demonstrating that tumoral ECM composition and density influence interstitial pressure. Interestingly, the injected dye did not reabsorb into blood vessels as expected perhaps due to the lack of blood flow observed around the area of injection. 24 In sum, the corrupted tumor-associated vasculature can protect the cancer from antitumor chemotherapeutic agents.
With our new understanding of the tumor as a complex organ, the current focus is on targeting the supportive tumor stroma. Based on observations that abnormal tumor vessels present difficulties in drug delivery to the tumor, efforts have been made to “normalize” tumor blood vessels in order to make for a more efficient delivery of antitumor drugs. By using neutralizing antibodies against VEGFR-2, hyperpermeable vessels can be “normalized”, and drug access to cancer cells can be improved. 23 Studies in mice have shown that VEGFR2-neutralizing antibodies are able to correct leaking vessels, restore a more normal VBM, decrease tumor interstitial pressure, and increase tumor oxygenation.27,28 Similar effects are seen in human tumors after treatment with bevacizumab (also known as Avastin [Genentech Inc., South San Francisco, CA], an antibody against VEGF) in coordination with chemotherapy and radiation. 29 Since VEGF-targeted therapies have been shown to have limited efficacy, the concept of targeting stromal cells in general has been brought to the forefront. 30 Guidance molecules including semaphorins, ephrins, netrins, and slit, well known to influence neuronal guidance, also have a role in vascular remodeling. 31 More specifically, semaphorins interact with Neuropilin-1 (NRP1) receptors to inhibit migration of endothelial and tumor cells alike. It was recently demonstrated that targeted and systemic delivery of Sema3A in a mouse tumor model abrogates tumor cell migration and metastasis as well as tumor angiogenesis. 32 By targeting the endothelium, the Sema3A starves the tumor for oxygen and nutrients and concomitantly prevents the tumor cells from migrating to seek out a new source of sustenance. The dual effect of this therapy has great potential. Moreover, understanding the various signaling molecules can provide further opportunities to effectively suppress the multiple survival pathways exploited by tumors.
Operational Signaling Pathways in Tumor Angiogenesis
In the last two decades, much has been learned about the tumor vasculature. As the tumor expands, growth factors secreted by tumor cells mediate the induction of angiogenesis and control the inflammatory infiltrate. Multiple ligand-receptor complexes have been associated with tumor angiogenesis. The network of growth factors and receptors during tumor growth is indeed similar to the one operative in developmental angiogenesis and during angiogenesis-driven inflammation33,34 (Fig. 3). Nonetheless, the lack of cross-regulatory control with the host tissue and the misregulation of downstream signals result in an aberrant vascular supply, one that is frequently inefficient with irregular flow patterns and abnormal delivery of oxygen and nutrients.16,17
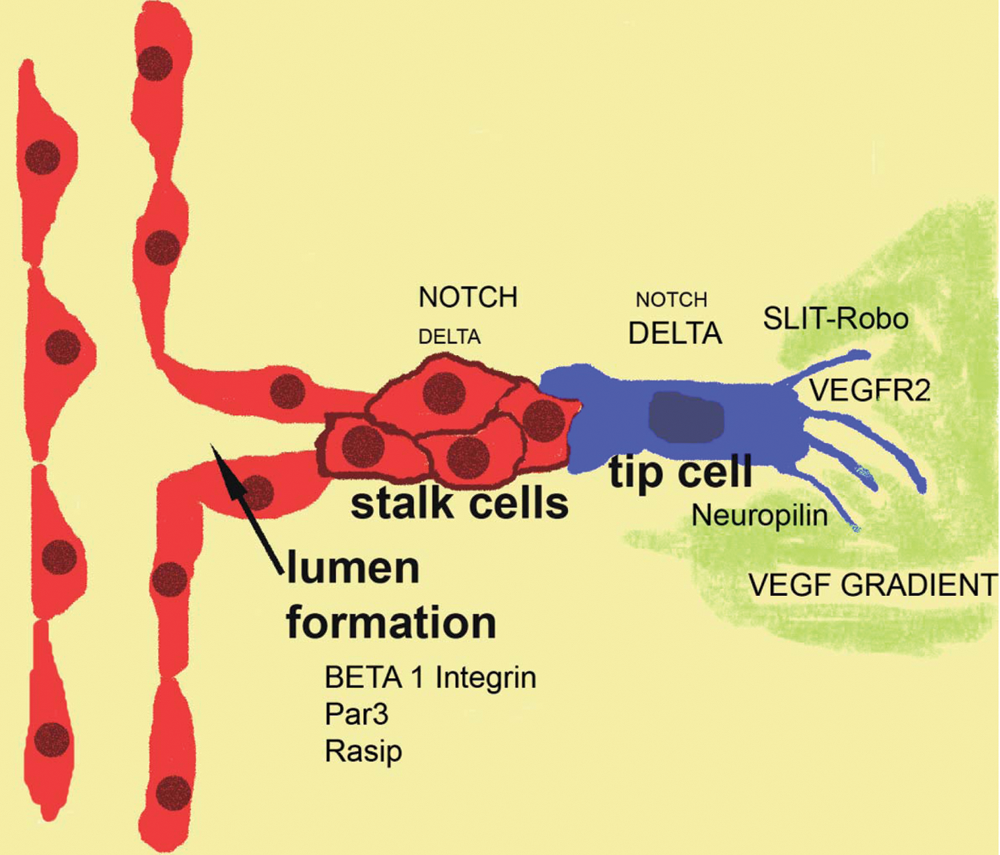
Signaling pathways in angiogenesis. The key cellular events associated with tumor angiogenesis are very similar to those described in developmental angiogenesis. A vascular sprout is composed of stalk cells and tip cells. The distinction between stalk and tip is conveyed by Notch signaling, which is higher in the stalk than in the tip cell. Tip cells received signals from a gradient of VEGF and from the Slit-Robo pathway.
To date, the following ligand-receptor signaling networks have been identified as contributors to tumor angiogenesis: 1) VEGF ligand with receptors VEGFR-1, VEGFR-2, and NRP1; 2) angiopoietin ligands with the Tie receptors; 3) Delta-like and Jagged ligands with Notch receptors; 4) Ephrin ligands with the Eph receptors; and 5) Slit ligand with the Robo receptors. Combined, the circuitry of growth factors and the respective receptors mediates the proliferation and migration of endothelial cells and the organization of vascular networks that feed the tumor and provide avenues for metastatic spread. 35 Stimulation of the endothelium by growth factors is critical to attain two of the hallmarks of cancer: angiogenesis and metastasis. 1 Thus, much effort has been placed in the suppression of tumor angiogenesis via targeting specific growth factors and receptors.36,37
Differential Effects of Soluble Versus ECM-Bound VEGF-A on Endothelial Cells
As a tumor outgrows the oxygen diffusion gradient, it either co-opts existing vasculature or induces angiogenesis. Prolyl hydroxylases (PHDs) allow cells (tumor and stromal) to sense the oxygen levels in the environment. 38 Specifically, under normoxic conditions, PHDs use the abundance of oxygen molecules to hydroxylate HIF transcription factors. Hydroxylated HIF associates with VHL (von Hippel-Lindau) and becomes targeted for degradation by the proteasome. Conversely, under hypoxic conditions, the HIF2α transcription factor in endothelial cells is able to induce the expression of target genes like VEGF-A (henceforth, referred to as VEGF).
VEGF is secreted in multiple isoforms, as a result of alternative splicing of a single gene, with VEGF164 being the most common. 39 Every isoform retains the receptor-binding domain encoded by exons 2 to 5 but differ in their carboxy-terminal end (encoded by exons 6a, 6b, 7, and 8). These differences alter their respective abilities to bind to the ECM and to the nonenzymatic single-pass transmembrane proteins Neuropilin-1 and -2 (NRP1 and 2). In addition to splicing events, these changes can be imposed by posttranslational proteolytic cleavage of the matrix-binding VEGF C-terminus, creating soluble forms. MMP and plasmin processing of VEGF generates 113 and 110 amino acid receptor–binding products, respectively.40,41 VEGF matrix associations increase the stability of the VEGF molecules in vitro. 42 More importantly, association of VEGF with NRP1 and the ECM confers different VEGFR2-mediated downstream signaling compared to soluble VEGF.42,43
Endothelial cells respond to the VEGF ligand through 2 receptor tyrosine kinases: VEGFR-1 and VEGR-2 (VEGF receptor-1 and -2, respectively). Most of the proangiogenic responses are conveyed by VEGFR-2, while VEGFR-1 is thought to act as a decoy modulator. Upon VEGF dimer-ligand binding, VEGFR-2 homodimerizes and induces cross-phosphorylation. Active VEGFR-2 sparks downstream signaling that can induce endothelial cell proliferation, migration, and vascular permeability. Soluble VEGF signals differently than matrix-bound VEGF. Mice expressing only the soluble VEGF isoform 120 have less vascular branching during embryogenesis, and more than half succumb to death as neonates. 44 Similarly, blood vessels induced by recombinant VEGF113 (made to mimic MMP-cleaved VEGF) had dramatically less branching than those induced by VEGF164. In contrast, VEGF that cannot be released from the matrix (VEGF Δ108-118) resulted in highly branched vasculature with a high density of thin vessels. 41 Similar findings were obtained using tumor cells that overexpressed VEGF113, 164, or Δ108-118. 41 These data demonstrate that soluble VEGF appears to be responsible for increasing the girth of vessels, while bound VEGF signaling influences vessel branching.
Recently, we have shown that the difference in human umbilical vein endothelial cell response to bound versus soluble VEGF can be explained by divergent downstream signaling pathways. 42 Both soluble and collagen matrix–bound VEGF can bind VEGFR-2 through the amino acids encoded by VEGF exons 2 to 5 and activate the receptor as measured by tyrosine 1175 phosphorylation. However, several differences exist in the nature of the signal and downstream activation. In particular, prolonged phosphorylation of VEGFR-2 at tyrosine 1214 is specifically found when VEGF is bound to matrix. Both soluble and collagen-bound VEGF can lead to phosphorylation of the secondary messengers p38MAPK and Akt; but collagen matrix–bound VEGF leads to prolonged p38MAPK activation, while soluble VEGF causes prolonged Akt activation. Interestingly, matrix-bound VEGF stimulates VEGFR-2 clustering and association with β1-integrin at focal adhesions. Inhibition of β1-integrin dampens the prolonged phosphorylation at VEGFR-2 Y1214, p28MAPK phosphorylation, and the levels of internalization of VEGFR-2. This implies that collagen-bound VEGF bridges the β1-integrin association with VEGFR-2 and that this interaction is responsible for the divergent signaling pathways. Thus, association of VEGF with the ECM brings VEGFR-2 in proximity to β1-integrin, which recruits an additional set of secondary messengers to be activated. These data suggest a mechanism for the differential cellular response to soluble versus bound VEGF. Alternative to or in addition to this pathway is the signaling pathway downstream of the VEGF coreceptor Neuropilin-1, which also binds to this C-terminal domain of VEGF.
VEGF/Neuropilin Signaling
The C-terminal domain of VEGF, encoded by exons 7 and 8, mediates binding to the nonreceptor tyrosine kinase Neuropilin-1 (NRP1).45-47 The role of NRP in vascular development is underscored by the observation that NRP1 null mice die of cardiovascular and neuronal defects between E12 and E13.5. 48 Neuropilin-1 and -2 are transmembrane glycoproteins expressed in multiple cell types including neurons, cancer cells, smooth muscle cells, and endothelial cells. 49 NRPs are capable of binding at least 2 different ligands: the semaphorins, which inhibit cell migration, and the growth factors like VEGF, PDGF, and HGF that promote cell migration. 50 Because NRP1 and 2 are expressed on both tumor and endothelial cells, it has been demonstrated that Sema3A can be used to target cancer with a dual approach. 30
The interacting region of NRP with VEGF is distinct from that of semaphorins, and it is mediated via its b1b2 extracellular domains.51,52 NRP1 has a short cytoplasmic tail with a single PDZ- binding domain, but it contains no inherent enzymatic activity, and its ability to signal on its own is not clear at this point. 49 For this reason, NRPs have been considered as coreceptors for VEGF rather than independent receptors. Indeed, NRPs have been shown to enhance VEGF interaction with VEGFR-2. 53 Despite this long-held assumption, the NRP1 cytoplasmic domain is capable of initiating endothelial cell migration. 54 Intracellularly, Synectin/GIPC1/NIP binds to the NRP1 PDZ motif using yeast 2-hybrid studies, and it is necessary for NRP1-mediated endothelial cell migration.55,56 Further downstream, P130Cas phosphorylation mediates VEGF-induced NRP1-dependent migration of endothelial cells. 57 A recent study pinpointed functional relevance to amino acids Y297 and D320 in NRP1. These residues, located in the b1b2 domain, are critical for VEGF binding to NRP1 and necessary for VEGF stimulation of endothelial cell migration. 58 In contrast, inhibition of VEGF binding to NRP1 had no effect on VEGFR-2–mediated endothelial cell proliferation and permeability. The emerging theme is that NRPs are relevant to the migratory function initiated downstream of VEGF signaling.
Small molecule inhibitors against NRP1, as well as NRP1-targeting antibodies, have shown potential in cancer therapy. In particular, the small molecule EG00229, a VEGF exon 7 and 8 mimetic, is able to compete for VEGF binding to NRP1 and inhibit VEGF-induced endothelial cell migration.57,59 Coupled with targeted delivery, this molecule holds clinical promise. Along the same lines, an antibody aimed at disrupting the interaction between NRP1 and VEGF yields reduction in pericyte coverage of tumor vessels. 60 Furthermore, simultaneous anti-NRP1 and anti-VEGF antibody treatment of tumor xenograft models resulted in inhibition of tumor volume and prolonged survival compared to either agent alone. The authors argue that the removal of pericytes by targeting NRP1 makes the vessels more vulnerable to anti-VEGF treatment.
Notch Signaling Determines Tip Versus Stalk Cell Fate in the Vascular Sprout
Notch signaling regulates the initial process of vascular sprouting by establishing critical differences between leading (tip) and following (stalk) cells. The Notch pathway has been well established as a regulator of cell fate and homeostasis in multiple settings including branching morphogenesis in tracheal sprouting and neuronal cell fate.61,62 Interestingly, sprouting angiogenesis has commonalities with both tracheal sprouting and neuronal development, including sensing and responding to environmental chemotactic and inhibitory guidance cues.63,64
Notch receptors and ligands are cell-surface single-pass transmembrane proteins that can engage in juxtacrine or autocrine signaling. 65 In mammals, there are 4 Notch receptors (Notch 1-4) and 5 ligands (Delta-like 1, 3, and 4 and Jagged 1 and 2). Within the secretory pathway, Notch receptors are cleaved by furin-like convertases into 2 fragments that later associate into a heterodimeric form. Thus, the mature cell surface receptor includes a membrane-spanning domain that is connected to an extracellular domain through noncovalent interactions. Upon ligand binding, the Notch receptor changes conformation of the juxtamembrane region, exposing a protease-sensitive domain to ADAM 10/17 that cleaves the extracellular portion of the receptor. The cleaved Notch extracellular domain–ligand complex is endocytosed by the ligand-expressing cell. The Notch intracellular membrane–tethered receptor is further cleaved by γ-secretase, a process that releases the Notch intracellular domain (NICD) from the cell membrane and enables its translocation into the nucleus. There, the NICD acts as a transcription factor, regulating the expression of various target genes like VEGFR-1, 2, and 3 in endothelial cells. 66
Notch activation by Dll4 (Delta-like 4) results in the formation of an angiogenic sprout with 2 distinct types of endothelial cells: tip and stalk cells. The tip cells lack Notch signaling and are responsible for sensing environmental cues through filopodia armed with VEGFR-2. 67 Upon VEGF binding to VEGFR-2, Dll-4 is upregulated via the PI3K/Akt pathway in arterial endothelial cells in vitro. 68 VEGF treatment increases Dll4 in mouse retinas ex vivo. 69 Tip cells and stalk cells closest to migrating front of the mouse retina express more Dll4 than do stalk cells, which express more Notch receptors than do the tip cells. 70 The tip endothelial cells at the leading edge of the vascular plexus receive the most exposure to VEGF and express the most Dll4, which in turn signal to the following endothelial cells to become stalk cells. Because Dll4 haploinsufficiency and pharmacological blockade of Notch signaling (by DAPT treatment) lead to more branching morphogenesis in the developing retina, the default phenotype would appear to be that of a tip cell. 70 Confocal imaging of developing mouse retinas shows that while tip cells are responsible for branching morphogenesis, the stalk cells form the blood-perfused tubes. 67 To accomplish this task, stalk cells must rapidly proliferate to fill in the plexus behind leading tip cells. In fact, the developing mouse retina shows a lack of proliferation in tip cells, in contrast to profuse proliferation by the stalk cells. 67
The Dll4-Notch cascade also regulates tip/stalk cell specification in tumor angiogenesis. Interestingly, several groups have shown that inhibiting Dll4 signaling paradoxically results in smaller tumors. Injection of Dll4 inhibitory antibodies into several xenograft mouse models yields smaller tumors than control-treated animals. 71 The mechanism by which tumor size was reduced lies in the observation that while there appeared to be more endothelial cells present in the tumor, they were unproductive and unable to form perfused vessels. Furthermore, while the vasculature of tumors overexpressing Dll4 had increased vessel density, they exhibited increased hypoxia due to insufficient vascular perfusion. 72 Given that information, the possibility of targeting Dll4 as a modality of cancer therapy has been explored in preclinical models. 73 However, because Notch signaling is involved in many cellular processes, concerns related to side effects are high. Furthermore, the concept of increasing the number of endothelial cells (tip cells) within a tumor is worrisome. While the presence of the Dll4 antibody would block their intrinsic ability to organize patent vascular tubes, it is likely that a short suppression of therapy might enable the formation of a vascular network, which would no longer be suppressed by antibodies. Additional experiments that explore the consequences of interruption of treatment are warranted.
Angiopoietin/Tie2 Signaling Maintains Vascular Quiescence
VEGF/VEGFR-2 stimulates angiogenesis, and the Dll4/Notch signaling axis controls endothelial tip/stalk cell fate promoting angiogenic growth. Nonetheless, functional vessels require the establishment of a quiescent phenotype to stabilize the new vascular network. Active cell signaling is needed to squelch further branching morphogenesis and to maintain homeostatic vessel integrity. Thus, after the establishment of a vascular plexus, a maturation process follows and it includes enhancement of tight junctions, secretion of a basement membrane, and recruitment of perivascular cells. For arteries and veins, this means recruitment of smooth muscle cells; for capillaries and small venules, it represents recruitment of pericytes. 74 Perivascular (mural) cells join the outer vessel wall through the stimulatory action of PDGFB/PDGFβ, HB-EGF/EGFR/HER2, HGF/c-Met, seratonin, sphingosine-1 phosphate, and TGF-β signaling.74-78 The recruitment of mural cells has been studied during development, but it is also relevant to tumor angiogenesis.79,80 In tumor vessels, the relative density and degree of adhesion of pericytes have been associated with alterations in response to therapy. The greater the number of pericytes and their degree of attachment to the endothelium, the more difficult it is to induce vascular regression. Furthermore, perivascular cells signal to the endothelium to maintain vessel quiescence and regulate permeability. Both perivascular paracrine and endothelial autocrine Tie/angiopoietin (Ang) signaling have been shown to influence quiescence and homeostasis of the vascular plexus.
The Tie/Ang signaling system comprises 2 Ang glycoprotein ligands (Ang-1 and -2) and 2 Tie tyrosine kinase receptors (Tie1 and 2). Tie1 and 2 are mainly, but not exclusively, expressed by endothelial and hematopoietic cells.81-83 Perivascular cells are the main producers of Ang-1, which once secreted is incorporated into the ECM. 74 Ang-2 is found mainly in endothelial cells, where it is stored intracellularly in Weibel- Palade bodies. 84 Perivascular Ang-1 has been shown to maintain quiescence by promoting endothelial cell survival, inhibition of vascular permeability, inhibition of proinflammatory signaling, and promotion of vascular maturation. 85
While Ang-1 signaling through Tie2 is thought to have an agonistic effect, the signaling outcome depends on how the ligand is presented to the receptor. Ang-1/Tie2 binding induces distinct signaling complexes when presented in cell-cell versus cell-matrix contexts.86,87 Endothelial cells engaged in cell-cell contacts show Tie2/Ang-1 in transhomotypic complexes. In contrast, endothelial cells that are not confluent or forming cell-cell connections showed Tie2 in association with the ECM. These distinct contextual localizations of Tie2 beget strikingly different signaling cascades. While cell-matrix Ang-1/Tie2 interactions lead to Erk/MAPK pathway stimulation, the cell-cell homotypic Ang-2/Tie2 complexes stimulate PI3K/Akt signaling. Further work demonstrated that Ang-2/Tie2 complex signaling leads to Dll4 expression via AKT-dependent activation of β-catenin and subsequent vessel maturation via basement membrane deposition. 78 Dll4 signaling via Notch induces the tip cell phenotype. Also contextual, the outcome of Ang-2 signaling through Tie2 is framed by the activation state of the endothelium. For example, Ang-2 activity destabilizes quiescent vasculature by competing with Ang-1 for Tie2 binding. 88 It has also been shown to destabilize the endothelium through an intracrine feedback loop. 89 Conversely, Ang-2 stimulation leads to the phosphorylation and activation of Tie2 and the inhibition of vessel leakage. 90 Less is known about the Tie1 receptor, but studies have shown that it can be cleaved to generate a soluble form 91 and that it can interact with and modulate the signaling of Tie2. 92
The Ang-2/Tie2 downstream signaling cascade has a role in cancer progression through activation of the endothelium. Interestingly, Tie2 is also expressed in multiple tumor cell types including Kaposi sarcoma, cutaneous angiosarcoma, melanoma, breast cancer, non–small cell lung cancer, hepatocellular carcinoma, prostate cancer, hemangioma, and astrocytoma. 93 Tumor-secreted Ang-2 can destabilize the endothelium and promote more branching morphogenesis. A special class of TAMs, named Tie2-expressing macrophages (TEMs), are able to home to tumors due to the local upregulation of Ang-2. These TEMs nurture tumor angiogenesis 94 and promote endothelial cell survival. 95 Accordingly, efforts are being made to target the Ang-2/Tie2 axis to suppress tumor angiogenesis by targeting these cells and the endothelium as well. 30 In particular, anti–Ang-2 monoclonal antibodies and the pharmacokinetically improved peptide-antibody conjugate called CovX-Bodies have been successful in preclinical trials.96,97 Furthermore, one such antibody, AMG386, has shown promise in ovarian cancer during phase II clinical trials. 98
Ephrin/Eph Axis in Arterial/Venous Patterning and Angiogenesis
Ephrin ligands and their Eph receptors generally provide repulsive signals for neurons. 99 They are divided into 2 classes, A and B, based on similarities in structure and binding affinities. 100 For example, class A Eph receptors typically bind all class A ephrin ligands and the same for class B receptors and ligands. However, there are some exceptions: EphA4 binds both class A and class B ephrins, while ephrinA4 binds both class A and class B receptors. 100 The Eph receptors are tyrosine kinases that are activated upon ephrin ligand binding. 101 The ephrin ligands are either GPI anchored (class A) or transmembrane (class B) cell-surface proteins. The Eph/ephrin receptor is unique, as it can initiate both forward and reverse signaling and, like Notch signaling, results in ligand/receptor endocytosis. Upon receptor/ligand binding, the ligand is cleaved from the cell surface. When the Eph receptors are endocytosed, the ligand goes along for the ride. 102
The contribution of Eph/ephrin signaling to vascular morphogenesis varies depending on the stage of vascular development, receptor class, and cancer type. Evaluation of reporter-transgenic mice showed EphB4 expression in veins and ephrinB2 in arteries.103,104 Homologous inactivation studies have shown that both EphB4 and ephrinB2 are critical for vascular development, as ablation of either gene causes embryonic lethality before E11.5.105-107 EphrinB2 expression is controlled by microenvironmental cues like VEGF exposure, smooth muscle cell association, and arterial flow stress.108,109 Given this and the fact that ephrinB2 and EphB4 tend to result in repulsion, it has been suggested that they define the vascular border. In addition, endothelial studies under culture conditions have revealed that activation of ephrinB2 by EphB4-Fc prevents migration and cell adhesions. 110 Additionally, ephrinB2/EphB4 appears to regulate the size of arteries and veins. 111 Furthermore, studies have demonstrated that ephrinB2 reverse signaling has a role in vessel maturation by influencing perivascular associations with the endothelium.112,113 Less is understood about the class A EphA2/ephrinA1 receptor ligand pair in developmental angiogenesis. However, ephrinA1 is expressed at sites of developmental angiogenesis, and soluble EphA2-Fc has been shown to inhibit endothelial cell migration, sprouting, survival, and corneal angiogenesis induced by VEGF.114,115
Disregulation of the ephrin/Eph molecules is observed in a wide variety of cancers. 116 Furthermore, both the class A ephrinA1 and EphA2 receptors are expressed by the endothelium and by tumor cells.117,118 Interestingly, hypoxia (a common feature of cancer) induces increased mRNA and protein levels of EphB4, ephrinB2, EphA2, and ephrinA1 in mouse skin. 119 Evidence that the ephrinA1/EphA2 axis plays a role in tumor angiogenesis lies in studies that show that tumors from EphA2 null mice are smaller and less vascularized.120,121 In addition, injection of soluble EphA2 and EphA3-Fc chimeras into tumor-bearing mice was able to inhibit tumor growth and angiogenesis.122-124
Two recent studies revealed a role for ephrinB2 in VEGFR-2 and VEGFR-3 endocytosis with consequences to developmental and tumor angiogenesis.125,126 Because VEGFR endocytosis is crucial for signaling, loss of ephrinB2 results in less VEGF receptor endocytosis, lower downstream signaling, and decreased tip cell filopodial extension. The net result is lower sprouting in the retina and a decrease in vessels permeating tumors in ephrin-deficient mice. Furthermore, injection of soluble EphA4 extracellular domains into mouse xenograft models of cancer results in reduced tumor size and tumor angiogenesis.127-129 In addition to their role in tumor angiogenesis, the majority of the studies have focused on the conflicting roles of Eph/ephrin in various types of tumor cells, in some instances promoting and in other times inhibiting tumor cell survival and metastasis. 116 In sum, further work should be done to tease out the nuances of Eph/ephrin signaling in cancer before using it as a target for cancer therapy.
Slit/Roundabout in Blood Vessel Guidance
Another group of neuronal signaling molecules recently found to modulate developmental and tumor angiogenesis is the Slit ligand/Roundabout (Robo) receptor duo. Mammals express 3 Slit ligands (Slit-1, -2, and -3) and 4 Robo receptors (Robo 1-4), which contribute to the morphogenesis and physiology of a wide variety of tissue types in addition to neurons. 130 Robo1 and Robo4 are the main Robo receptors in angiogenesis. Because our understanding of Robo/Slit signaling in angiogenesis is recent and limited, there exists some controversy surrounding the subject. However, it is clear that as in neuronal development, the Slit/Robo system participates in blood vessel guidance. In addition to its effects in developmental angiogenesis, Robo1 was shown to have a role in tumor neovascularization. 131 In a tumor context, Slit-2 is expressed by tumor cells, while the ligand, Robo1, is present in the endothelium. Slit-2 was shown to induce endothelial cell migration and tube formation in a PI3K-dependent manner.
Discovered within the last decade, Robo4, also known as magic Roundabout, is the only Robo receptor exclusively found in the endothelium. 132 Efforts made to further understand the role of Robo4 in blood vessel homeostasis have yielded conflicting results: some studies conclude that Robo4 activation inhibits endothelial cell migration, while others claim that it actually has promigratory effects. Evidence for Robo4 acting to induce endothelial cell migration lies in the findings that soluble Robo4-Fc inhibits both in vitro and in vivo angiogenesis by competing with the ligand for endogenous Robo4. 133 Interestingly, this group also argues that Slit-1 to -3 do not bind the Robo4 receptor, given measurements of weak interactions between purified forms of the two using the BiaCore system (Amersham Biosciences, Pittsburgh, PA). This finding is possibly due to the use of purified recombinant protein interactions in the absence of heparin, which is known to aid in the interaction between Robo and Slit.134,135 Another investigation into the promigratory role of Robo4 in endothelial cells demonstrates that Slit-2 binds to a Robo1/Robo4 heterodimer, which recruits WASP, leading to Robo4-dependent filopodia formation. 136 Alternatively, there are also data demonstrating that Robo4 inhibits angiogenesis and stabilizes the vasculature. 137 First, the authors showed that Robo4 is expressed predominantly in stalk cells of developing retinas. Next, they used Robo4 knockout mice to show that Robo4 inhibits VEGF-induced migration, tube formation, and permeability. Finally, they show that Slit-2 prevents Robo-mediated oxygen-induced retinopathy. The same group went on to illustrate that Slit/Robo signaling inhibits Arf6, an effector molecule downstream of VEGFR-2, which controls VEGF-induced endothelial cell migration, angiogenesis, and vascular permeability. 138 Interestingly, inactivation of Robo4 in endothelial cells produces more VEGF, has higher levels of activated VEGFR-2, and has increased angiogenic capacity in a mammary fat pad transplant assay. 139 This study also identified smooth muscle cells as the source of Slit-2 and Slit-3 in the developing mammary fat pad, underscoring the relevance of the microenvironment in angiogenesis. Seeking to reconcile the different effects that Slit-2 has on Robo signaling, one group has presented evidence that suggests that ephrinA1 inhibits the normally proangiogenic effects of Slit-2–induced mTORC2-Akt-Rac signaling pathway by inhibiting Akt and Rac. 140 Unfortunately, this article does not address specific Robo receptors. In addition, recent data demonstrate that Robo4 binds and activates Unc5b, inhibits Src signaling downstream of VEGF, and prevents VEGF-induced hyperpermeability and angiogenesis. 141
Conclusions
The last 20 years of research in vascular signaling have revealed a significant degree of complexity and cross-talk between signaling systems. Not surprisingly, all of the essential players noted to be active in tumor angiogenesis are reiterations of those operational during developmental angiogenesis. Major strides in the last 3 years have been made in clarifying the contribution of “context” in endothelial signaling. Thus, soluble versus matrix-bound VEGF or cell-cell versus cell-matrix activation of Tie2 both result in distinct downstream activation pathways and offer altered cellular responses. In addition, the contribution of inflammatory cells, particularly with respect to the production of matrix metalloproteases and their ability to modify the stroma, cytokines, and responding receptors has also gained visibility. While not currently being considered in the clinic, combination therapies that target tumor macrophages with antiangiogenic therapy might offer significant benefits. Increased knowledge on the contribtions of Notch signaling and its ligands has propelled the generation of antibodies for pharmacological blockade of this pathway in cancer. These, now in clinical trials, will offer complementation to the current anti-VEGF therapy and clarify one of the current questions in the field, namely whether inhibition of multiple signaling arms would be more effective than single therapies.
The quest to gain additional depth in the mechanisms that regulate vascular growth in tumors will continue to expand and deliver new ideas for therapeutic exploration. However, a more effective follow-up on how therapy alters tumor vasculature is needed to effectively translate these therapies when resistance to the therapy surfaces in the clinic, as it has with Avastin. Future experiments and more integrated efforts on the evaluation of the therapy at the molecular level will further aid to refine targeted treatments in tumor biology.
Footnotes
Acknowledgements
The authors thank and acknowledge members of the Arispe laboratory, in particular, Dr. Carmen Warren and Ms. Courtney Domigan, for critical reading of the article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the Ruth L. Kirschstein National Research Service Award [GM007185] to Ms. Ziyad; and the National Institutes of Health [grant numbers RO1 HL085618 and RO1 CA126935] to Dr. Iruela-Arispe.