
Abstract
Background/Objectives
Allicin, a bioactive compound from garlic, exhibits antibacterial activity by targeting bacterial proteins, including type II topoisomerases such as DNA gyrase and topoisomerase IV enzymes exclusive to prokaryotes. This study aimed to investigate the antibacterial effects of allicin and its inhibitory mechanism on topoisomerase IV using both in vitro and in silico approaches.
Methods
In the in vitro experiments, the antibacterial activities of allicin, garlic extract, and ciprofloxacin were evaluated using agar diffusion, as well as determination of minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC). In silico analyses included consensus sequence-based structural modeling, molecular docking, and molecular dynamics simulation of DNA gyrase and topoisomerase IV subunit A in the presence of allicin and ciprofloxacin.
Results
In vitro results demonstrated that the allicin exhibited the most potent antibacterial activity (P ≤ 0.05), followed by garlic extract, while ciprofloxacin demonstrated relatively lower activity. Docking analysis revealed stronger interactions and higher binding affinity between allicin and topoisomerase IV compared to the other ligands. Molecular dynamics simulations further confirmed the enhanced stability of the allicin-topoisomerase IV complex. Additionally, genomic analysis was used to predict the conserved region of the protein and generate a refined structural model, which showed improved binding characteristics.
Conclusion
These findings suggest that allicin is a promising inhibitor of bacterial topoisomerase IV and may serve as the basis for developing new antimicrobial agents.
Keywords
Introduction
Garlic (Allium sativum), which belongs to the family Amaryllidaceae, shows antibiotic activity against bacteria, especially strains that are resilient to traditional medicines. 1 Garlic extract possess the ability to inhibit the growth of certain harmful bacteria, affirming its effectiveness as a natural antibacterial agent.2,3 In several species of Allium, allicin has been found to be highly concentrated and this finding was supported by heatmap result (Figure 1). 4 Previous studies stated that allicin significantly contributed to garlic's antibacterial effects, 5 and recorded higher antibacterial activities against Staphylococcus aureus than gentamicin. 6 Allicin can be produced by various methods, including biosynthesis and enzymatic reactions. 7 Through enzymatic reactions, the allicin is produced by converting cysteine to γ-glutamylcysteine, followed by the formation of alliin via the action of alliin synthase.8,9 When alliinase reacts with alliin, allicin is formed, containing a thiosulfinate functional group (-S(O)-S-), which contributes to its antibacterial activity. 10

Compounds Containing Sulphur are Shown in a Heatmap. The Sulphur Compounds With the Strongest Signals in Various Types of Garlic Were Allicin and Alliin. 4
DNA gyrase and topoisomerase IV are classified into bacterial type II topoisomerases which play important roles in DNA replication, transcription and segregation of chromosomes. 11 Bacterial type II topoisomerases only are present in the bacterial cell, making them viable targets for designing novel drugs to treat bacterial infection.11,12 DNA gyrase and topoisomerase IV have a similar tetramer conformation but are involved in different intracellular functions in bacterial cells for survival. 13 DNA gyrase contains 2 subunits of GyrA and GyrB while topoisomerase IV comprises 2 subunits of ParC and ParE. DNA gyrase is mainly involved in introducing negative supercoils into DNA and eliminating positive supercoils formed before DNA tracking systems, replication forks and transcription complexes. On the other hand, topoisomerase IV removes the DNA knots and decatenates the tangles produced during recombination and replication of DNA.13–15 Fluoroquinolones are the most common classes employed to inhibit the type II topoisomerase in the recent era. 16 However, the increased utilization of fluoroquinolones leads to the development of resistance in bacterial cells. Thus, novel antimicrobial drugs that target topoisomerase and do not cross-resistance with the current 6-fluoroquinolones are desperately needed.
Allicin can be utilized to inhibit various bacterial proteins, such as DNA gyrase. 17 A previous study also stated that allicin was a potential drug to defend against antibiotic-resistant bacteria by inhibiting the important proteins involved in promoting bacterial growth and drug resistance. The target proteins used in this research are demonstrated in Figure 2. Among these proteins, allicin had greater binding affinity with dihydrofolate reductase, filamenting temperature-sensitive mutant Z, DNA gyrase A and B. 18 As DNA gyrase and topoisomerase IV belong to bacterial type II topoisomerases, topoisomerase IV could become a potential target of allicin. However, the action of allicin on topoisomerase IV is still unknown. In this study, the effects of allicin on a novel target protein, topoisomerase IV subunit A was investigated and its inhibitory mechanism was unraveled through binding interactions and molecular mechanisms using bioinformatic tools. The genomic analysis was used to predict the conserved region of protein for modelling of a novel structure. This approach further improved the binding between the target protein with the desired ligands.

Docking Results of Allicin With Various Bacterial Proteins/Enzymes in a Heatmap. 18
Material and Methods
The allicin (ChemFaces-CAS: 539-86-6) was procured from a reputable company, while ciprofloxacin was obtained from Bioanalyse cE, Turkey. In the experimental phase, antimicrobial procedures were compared, involving garlic extract, allicin, and ciprofloxacin (a conventional antibiotic). The concentrations used for garlic extract and allicin were 10 μg/ml, 20 μg/ml, and 30 μg/ml, and for ciprofloxacin, it was 5 μg/ml. To evaluate the inhibitory activity, agar diffusion techniques, and concentration of minimal inhibitory and bactericide were employed to assess the growth inhibition of both Gram-negative and Gram-positive bacterial isolates. Additionally, the study included a structural analysis of the DNA gyrase A and topoisomerase IV subunit A. A protein-ligand docking was performed to study the interactions of allicin with the target proteins, with ciprofloxacin serving as controls. The goal was to determine the energy and affinity between the inhibitors and the target proteins. Furthermore, the stability, convergence, and binding free energy of the protein-ligand complexes were evaluated using molecular dynamics simulation (MDS). Figure 3 showed the experimental design of the present work.
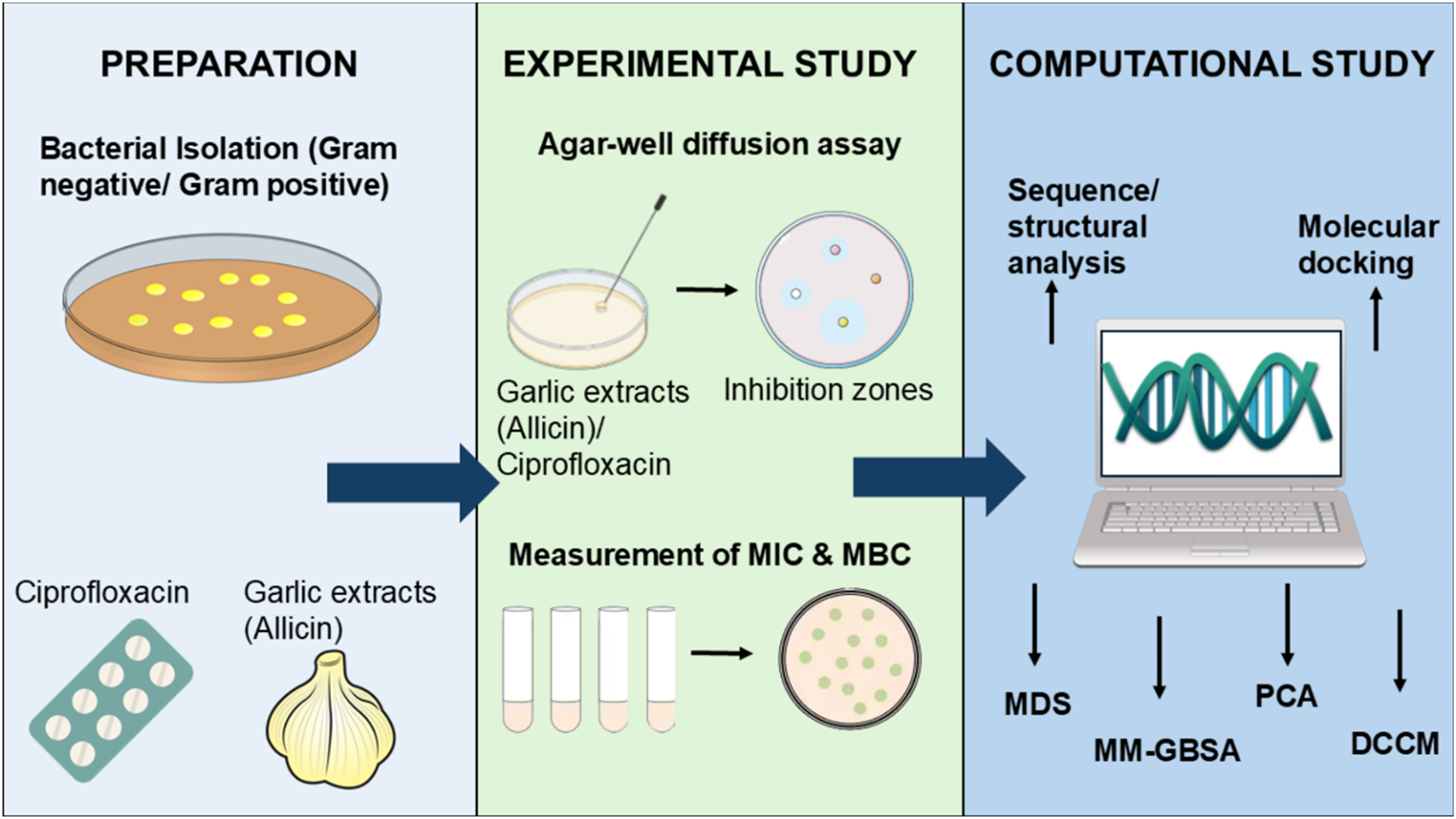
The Experimental Design of the Study.
Bacterial Isolates
The bacterial isolates that are listed in Table 1 were obtained from the Microbiology Laboratory at the College of Nursing, University of Babylon after being identified using the 16S rRNA method.
Gram-Positive and -Negative Bacteria.

Assessment of Antimicrobial Activity Using Agar-Well Diffusion Assay (In Vitro)
The procedure was according to previous studies by Hindi.19,20 A uniform bacterial suspension (containing 1.5 × 108 CFU / ml) was streaked onto the MuellerHinton agar surface using a cotton swab and the plate was air-dried for 5 −15 min. 8 wells with 5 mm diameter were cut from media using cork borer, and proper amounts of garlic extract, allicin, and ciprofloxacin were added into the wells. After 24 h of incubation at 37 °C, the zones of inhibition for all isolates were identified and measured by measuring in millimeters (mm). The steps were repeated for all tested bacteria and performed in triplicates.
Antibacterial Activity Assay
According to the study by Forbes, 21 the agar-disc diffusion method was utilized to evaluate the antimicrobial activity of garlic extract, allicin, and ciprofloxacin with the tests performed in triplicates.
Determination of the Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
The MIC and MBC of garlic extract, allicin and ciprofloxacin were determined by the two-fold agar dilution susceptibility approach. Based on the study by Miles and Amyes (1996), 22 the dilution ranges of MIC and MBC for each solution (μg/mL) were determined. The dilutions of garlic extract, allicin and ciprofloxacin in liquid Muller-Hinton agar were prepared as described by Ericsson and Sherris. 23 The diluted solutions were added into the molten Muller-Hinton agar media equilibrated in a water bath at 45–50 °C for mixing. The mixed solutions were poured into Petri dishes and solidified at room temperature. 1 μL of the adjusted inoculum (compared with the turbidity of 0.5 McFarland standards) was streaked onto the agar surface using a loop. After leaving the inoculated plates at room temperature for 30 min, the incubation of the plates was performed at 35 °C for 16 to 20 h. The MIC was defined as the minimal amounts of the antimicrobial agent used to suppress bacteria while the MBC was recorded as the lowest quantity of the antimicrobial solution employed to eliminate 99.9% of the bacteria. The MIC values were compared with the breakpoints suggested by NCCLS (2002). 24 Triplicates of MIC and MBC for each solution on bacterial isolates were determined, and their averages were recorded.
Sequence Analysis for Gyrase and Topoisomerase IV
The FASTA sequences for DNA gyrase and DNA topoisomerase IV for different bacterial strains were first retrieved from Uniprot. 25 A multiple sequence alignment was performed using Clustal Omega for DNA gyrase subunit A and DNA topoisomerase IV subunit A. 26 The alignment included the sequences of 14 species obtained from uniport: Pseudomonas aeruginosa, Acinetobacter baumannii, Enterococcus faecalis, Escherichia coli, Klebsiella aerogenes, Klebsiella pneumoniae, Proteus mirabilis, Proteus vulgaris, Pseudomonas fluorescens, Salmonella typhi, Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus pneumoniae, Streptococcus pyogenes, the accession IDs are given in the table in S1 Table. EMBOSS cons was employed to generate a consensus sequence for DNA gyrase subunit A and DNA topoisomerase IV subunit A from a multiple sequence alignment. 27
Tertiary Structure Prediction of DNA Gyrase and Topoisomerase IV
The structures of topoisomerase IV and gyrase were predicted using consensus sequence through several webservers such as Phyre 2 (http://www.sbg.bio.ic.ac.uk/∼phyre2/html/page.cgi?id=index), 28 SWISS-MODEL (https://swissmodel.expasy.org/), 29 Sequence Similarity Search (https://www.ebi.ac.uk/jdispatcher/sss/fasta) 30 and AlphaFold Colab (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb). 31 The PDB files of the modelled structures were then submitted to the Structural Analysis and Verification Server (SAVESv6.1) (https://saves.mbi.ucla.edu/) for validation. Their quality was assessed through ERRAT, 32 VERIFY3D, 33 and PROCHECK. 34 The PROCHECK was utilized to determine the model's stereochemical quality. The ERRAT was additionally employed to analyze the overall model quality whereas VERIFY3D determined the compatibility between the tertiary conformation and primary amino acid sequence of the predicted models. 35 The predicted model was sent to GalaxyRefine (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) 36 for refinement if the percentage of the Ramachandran plot generated was below 90%. The predicted models with the highest quality were chosen and their structures were visualized using PyMOL (https://pymol.org/). 37
Molecular Docking
Protein-ligand docking was conducted on AutoDock Vina Version 1.1.2 (https://vina.scripps.edu/downloads/) 38 by docking the ligands that included allicin as well as ciprofloxacin into topoisomerase IV and gyrase to measure the binding affinity. The smiles files of allicin (ID: 65036) and ciprofloxacin (ID: 2764) were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). 39 The molecular structures of allicin and ciprofloxacin were modelled and optimized using Avogadro2 (https://avogadro.cc/). 40 Next, the PDB files of topoisomerase IV, gyrase, allicin, and ciprofloxacin were changed and saved into PDBQT file format for docking preparation. Molecular docking of topoisomerase IV with ligands was conducted within a grid box with a size of 36 × 26 × 34 Å. The X, Y, and Z coordinates for the center of the grid box were −0.75, −2.71 and −0.81, respectively. For molecular docking between ligands and gyrase, the size and coordinates of the grid box were 30 × 27 × 27 Å and 3.59 × 12.51 × 0.98, respectively. For each effective search, the default parameters were utilized, except the number of modes was set to 1000. The docked conformations having the lowest binding free energies were selected and their interactions were visualized using Biovia Discovery Studio Version 2024 (https://discover.3ds.com/discovery-studio-visualizer-download). 41
Molecular Dynamics Simulation
Desmond, Schrödinger LLC was utilized to conduct Molecular dynamic simulations for 100 nanoseconds. Then, the Protein Preparation Wizard of Maestro was employed to preprocess the receptor–ligand complex, including optimization and minimization of the complex. The preparation of all the systems was performed using the System Builder tool. In the simulation, Transferable Intermolecular Interaction Potential 3 Points (TIP3P), which is a solvent model with an orthorhombic box, and the OPLS 2005 force field were used.42,43 The counter ions were introduced to neutralize the models during simulation. To mimic physiological conditions, 0.15 M sodium chloride (NaCl) was added.44,45 The NPT (number of particles, pressure, and temperature) ensemble with 310 K temperature and 1 atm pressure was selected for the entire simulation. Before the simulation, the relaxation of the models was conducted. Every 100 ps, the trajectories were saved for analysis. The stability of the simulation was verified through comparison among the protein and ligand's root mean square deviation (RMSD), root mean square fluctuation (RMSF), and ligand properties (radius of Gyration, solvent-accessible surface area (SASA), hydrogen bond, etc) over time. Desmond software with default parameters was utilized to conduct the complexes’ principal component analysis (PCA) and Dynamic Cross-Correlation Matrix (DCCM).
The final production run was conducted for 200 ns. To analyze the (PCA), Geo_measures v 0.8 was employed. 46 Geo_measures includes a robust library of g_sham and eigenvalues, which are depicted in a 3D graphic using the Python tool matplotlib. Each last processing run lasted for 200 ns. To assess the stability of the MD simulations, root mean square deviation (RMSD), radius of gyration (Rg), root-mean-square fluctuation (RMSF), and the number of hydrogen bonds (H-bonds) were determined.
Analysis of Binding Free Energy
The binding free energies of the ligand-protein complexes were determined using the molecular mechanics combined with generalized Born surface area (MM-GBSA) method. The Prime MM-GBSA binding free energy was calculated using the thermal mmgbsa.py Python script, analyzing the simulation trajectory for the past 50 frames with a 1-step sampling size. The binding free energy of Prime MM-GBSA (kcal/mol) was obtained by considering the additivity concept, which involves summing up each individual energy module. These modules include columbic, covalent, hydrogen bond, van der Waals, self-contact, lipophilic, solvation of protein, and ligand energies.
The equation used to calculate ΔGbind (binding free energy) is as follows:
Where:
ΔGbind represents the binding free energy,
ΔGMM is the difference between the free energies of the ligand-protein complexes and the total energies of the protein and ligand in their isolated forms,
ΔGSolv represents the difference in the GSA solvation energies between the ligand-receptor complex and the sum of the solvation energies of the receptor and the ligand in the unbound state,
ΔGSA denotes the difference in the surface area energies for the protein and the ligand.
Statistical Analysis
Data analysis was conducted using SPSS version 23, Bonferroni test 47 to determine significant differences between the garlic extract, allicin, and ciprofloxacin. The significance level was set at (P ≤ 0.05) to identify statistically significant results.
Result
Estimating Antimicrobial Activity
The results showed significant differences in the inhibitory effects of allicin compared to garlic extract and ciprofloxacin in all bacterial isolates at a significance level of (P ≤ 0.05). Additionally, ciprofloxacin demonstrated no activity against some Gram-negative bacteria. The significant concentration of allicin (10 μg/mL) completely inhibited all of the examined organisms, with the largest inhibition zone (48 mm) found against E. feacalis .While the garlic concentration (30 μg/mL) was recorded (38 mm) against E. feacalis. On the other hand, the results revealed that ciprofloxacin (5 μg/mL) had the lowest activity against bacteria. The maximum inhibition zone, which measured 15 mm, was shown to be inactive towards the majority of isolates. According to the findings, the allicin had the strongest benefits, whereas ciprofloxacin had fewer ones.
Additionally, the effects of allicin on microbes revealed that all bacterial isolates were quite sensitive to this mixture. Enterococcus feacalis was greatly sensitive to allicin than other bacteria, with a 48 mm zone of inhibition, followed by S. pneumoniae, K. pneumoniae, K. aerugenes, E. coli, P. mirabilis, P. fluorescence, and P. aeruginosa, with a range of inhibition zones were 38–47 mm (Table 2).
Garlic Extract, and Allicin by Agar Well Method and Ciprofloxacin by Disc Diffusion Method All Demonstrated Antimiriacrobial Activity (Inhibition Zones (mm)).

Determination of the Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
Based on Table 3, the allicin had the lowest values of MIC and MBC on bacterial isolates compared to garlic and ciprofloxacin. The MIC of allicin on Staphylococcus epidermidis and Enterococcus faecalis was 8 µg /mL which was the lowest among the bacterial isolates. The largest MIC value of allicin within bacterial isolates was 32 µg /mL which were the results on Pseudomonas aeruginosa and Pseudomonas flourscences. The MIC of allicin on other bacterial isolates was 16 µg /mL. The MBC of allicin on bacterial isolates was double the MIC of allicin. The MIC and MBC values of garlic on bacterial isolates were two-fold and four-fold the MIC values of allicin. The MIC and MBC values of ciprofloxacin on bacterial isolates were the greatest which had 512 µg /mL, except the MIC on Salmonella typhi, Escherichia coli spp., and Gram-positive bacteria which was 256 µg /mL. These MIC and MBC analyses were performed three times, and the average values were used.
Determination of MIC and MBC (µg /mL) for Garlic, Allicin and Ciprofloxacin on the Bacterial Isolates.

Sequence and Structural Analysis
The domain of a protein is the functional region that determines its function. The locations of the domain present in DNA gyrase and topoisomerase IV were identified, which both inhibitors bind within the domain region of both proteins. The conserved regions for each protein subunit A were determined through multiple sequence alignments (Figure 4C and Figure 4D). The phylogenic relationship of the gyrase and topoisomerase IV between all tested bacteria was shown in Figure 4A and Figure 4B.

Genomic Data of Gyrase and Topoisomerase IV. (A) Phylogenic Tree of Gyrase of All Tested Bacteria. (B) Phylogenic Tree of Topoisomerase IV of All Tested Bacteria. (C) Predicted Consensus Sequence of DNA Gyrase Across Different Bacteria. (D) Predicted Consensus Sequence of Topoisomerase IV Across Different Bacteria.
Tertiary Structure Prediction of DNA Gyrase & Topoisomerase IV
The validation results of topoisomerase and gyrase modelled by different webservers (before refinement) are shown in S2 Table and S3 Fig. to S13 Fig. (Supplementary file). Based on the results in the S2 Table, only the structures predicted by AlphaFold Colab contained the number of residues that were tallied with sequences of topoisomerase and gyrase. However, their percentages of PROCHECK were below 90%. Thus, the topoisomerase and gyrase structures predicted by AlphaFold Colab were refined using GalaxyRefine. After refinement, the results of ERRAT, VERIFY3D and PROCHECK (S1 Fig. and Fig. 5E) for the modelled topoisomerase IV were 96%, 95.38%, and 97.9%, respectively. Meanwhile, gyrase obtained 78.02% of the overall quality factor and 49.77% of residues having an averaged 3D-1D score that is more or equal to 0.1 (S2 Fig.). According to the Ramachandran plot (Figure 5F), 97% of gyrase residues fell within the most favoured regions, but gyrase also contained 0.6% of residues falling within the disallowed regions. The tertiary structures of topoisomerase IV and gyrase are shown in Figure 5A and Figure 5B. Topoisomerase IV was made up of 5 α-helices connected by loops whereas gyrase was made up of 2 antiparallel β-strands and 3 α-helices linked by loops. The structures of allicin and ciprofloxacin are shown in Figure 5C and Figure 5D.

Tertiary Structure Informations of Proteins and Ligands. (A) Tertiary Conformation of Topoisomerase IV Predicted by AlphaFold Colab. (B) Tertiary Conformation of Gyrase Predicted by AlphaFold Colab. (The α-Helices are Red in Colour, β-Strands are Yellow in Colour and the Loops are Green in Colour.) (C) Tertiary Structure of Allicin (Yellow Stick Form). (D) Tertiary Structure of Ciprofloxacin (Orange Stick Form). (E) Ramachandran Plots of Topoisomerase IV After Refinement. (Topoisomerase IV had 97.9% and 2.1% of Residues that Fell Within the Most Favored and Additional Allowed Regions.) (F) Ramachandran Plots of Gyrase After Refinement. (Gyrase had 97%, 2.4% and 0.6% of Residues Falling Within the Most Favored, Additional Allowed and Disallowed Regions, Respectively).
Molecular Docking Studies of Proteins Against Inhibitors
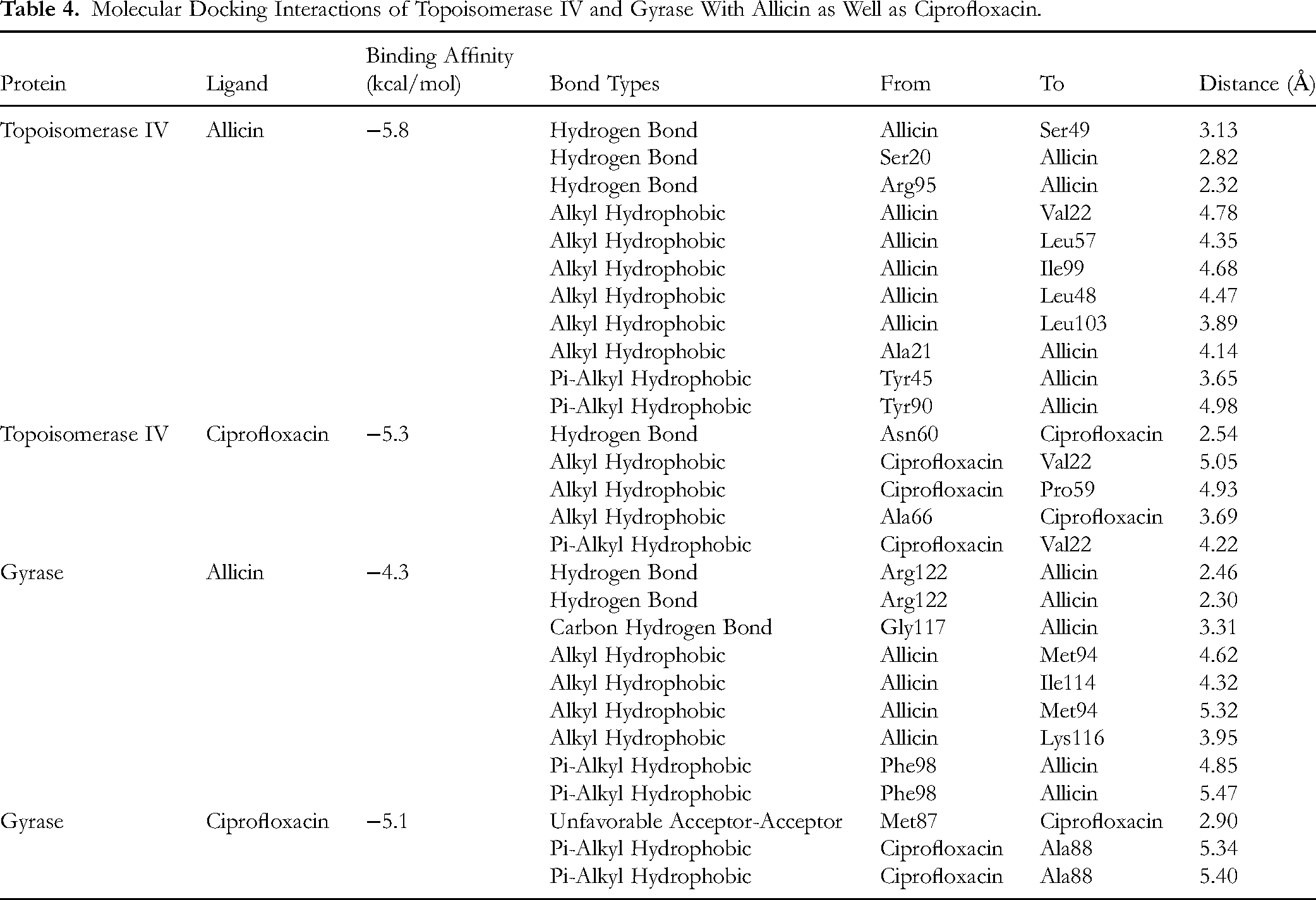
The binding affinities between proteins (topoisomerase IV and gyrase) with ligands (allicin and ciprofloxacin) were determined through molecular docking. Based on the findings, the binding affinity of Topoisomerase IV-Allicin was −5.8 kcal/mol. Allicin bound to topoisomerase IV at one end of the centre of the 3 α-helices (D8 to V22, G39 to T52, and R94 to G114). It formed 3 hydrogen bonds with Ser49, Ser20 and Arg95 of topoisomerase IV. There were 8 hydrophobic interactions formed between allicin and topoisomerase IV (Val22, Leu57, Ile99, Leu48, Leu103, Ala21, Tyr45, and Tyr90). In the docking between topoisomerase IV and ciprofloxacin, the ciprofloxacin was located near the α-helix formed by D8 to V22 and a loop (N53 to P68). The binding affinity between topoisomerase IV and ciprofloxacin was −5.3 kcal/mol. 1 hydrogen bond was formed with Asn60 and 4 hydrophobic interactions were formed between ciprofloxacin with Val22, Pro59, and Ala66 of topoisomerase IV. For gyrase, the molecular docking with allicin and ciprofloxacin were −4.3 kcal/mol and −5.1 kcal/mol, respectively. In the docking between gyrase and allicin, the allicin bounds to the space near the α-helix formed from residue K112 to E120 and the loop generated by resides G105 to N111. 2 hydrogen bonds (Arg122), 1 carbon hydrogen bond (Gly117) and 6 hydrophobic interactions (Met94, Ile114, Lys116 and Phe98) were formed in the Gyrase-Allicin complex. When ciprofloxacin was docked with gyrase, only 2 hydrophobic interactions formed with Ala88, and 1 unfavourable acceptor-acceptor interaction formed with Met87. The ciprofloxacin bounds to the space near the β-strand formed from residue A84 to T89 and the loop generated by resides N90 to T100. The molecular interactions between proteins and ligands are shown in Figure 6 and Table 4.

Molecular Surface View of Allicin and Ciprofloxacin With Target Proteins. (A) Topoisomerase IV-Allicin, (B) Topoisomerase IV-Ciprofloxacin, (C) Gyrase-Allicin, and (D) Gyrase-Ciprofloxacin. (Left Panel) (The Surfaces of Topoisomerase IV and Gyrase are Shown in Grey and Light Blue Colour While the Allicin and Ciprofloxacin are Shown in Yellow and Orange Stick Forms). 3D and 2D Interaction With Hydrogen Bonds and Other Non-Bonded Interactions. (Right Panel) (H: Hydrogen Bond; HP: Hydrophobic Interaction; UF: Unfavourable Bond).
Molecular Docking Interactions of Topoisomerase IV and Gyrase With Allicin as Well as Ciprofloxacin.
Molecular Dynamics Simulation (MDS)
To assess the stability and convergence of gyrase and topoisomerase IV with drugs, allicin and ciprofloxacin, extensive MDS were conducted. The root mean square deviation (RMSD) measurements during the 200 ns simulations indicated the stability of the conformations for both complexes. When the RMSD values fell within the acceptable range of less than 3 Å, it suggested that the complex had good convergence and stability throughout the simulation. The Cα-backbone's RMSD values of gyrase bound to allicin and ciprofloxacin were 21.0 Å and 24.0 Å (Figure 7 A1). Allicin-bound protein topoisomerase IV displayed an RMSD value of 7.0 Å, while the ciprofloxacin-bound protein exhibited an RMSD of 9.0 Å (Figure 7 A2). It indicated that allicin had greater binding affinity with the target proteins and formed a more stable complex during simulation.

MDS Analysis of 200 ns Trajectories of Inhibitors and Target Proteins. (A1) Cα Backbone RMSD of Gyrase+Allicin (Black), Gyrase+ Ciprofloxacin (Red), (A2) Cα Backbone RMSD of Topoisomerase IV+Allicin (Black) and Topoisomerase IV+Ciprofloxacin (Red). (B1) RMSF of Cα Backbone of Gyrase+Allicin (Black), Gyrase+Ciprofloxacin (Red), (B2) Cα Backbone RMSF of Topoisomerase IV +Allicin (Black) and Topoisomerase IV+Ciprofloxacin (Red). (C1) Rg of Gyrase+Allicin (Black), Gyrase+Ciprofloxacin (Red), (C2) Rg of Cα Backbone of Topoisomerase IV IV +Allicin (Black) and Topoisomerase IV +Ciprofloxacin (Red). (D1) Formation of Hydrogen Bonds in Gyrase+Allicin (Black), Gyrase+Ciprofloxacin (Red), (D2) Formation of Hydrogen Bonds in Topoisomerase IV+Allicin (Black) and Topoisomerase IV +Ciprofloxacin (Red). (E1) SASA in Gyrase+Allicin (Black), Gyrase+Ciprofloxacin (Red), (E2) SASA in Topoisomerase IV+Allicin (Black) and Topoisomerase IV +Ciprofloxacin (Red).
The RMSF plot indicated fluctuations with several spikes in the gyrase protein bound to allicin. The small spikes were at residues 15–20, 90–100, 170–175 and 210–217, indicating increased flexibility. When treated with ciprofloxacin, gyrase showed variability at the 40–50, 65–70 and 180–190 residue locations (Figure 7 B1). On the other hand, allicin-bound protein topoisomerase IV demonstrated minimal fluctuations with 2 small spikes at residues 70–80 and 110–120. The RMSF plot of the ciprofloxacin-bound protein showed a similar pattern but the spikes were located at different locations that were around residues 75–79 and 115–125 (Figure 7 B2). The flexibility of the Topoisomerase IV-Allicin was the lowest followed by Topoisomerase IV-Ciprofloxacin, Gyrase-Ciprofloxacin, and Gyrase-Allicin complexes.
The Cα backbone radius of gyration (Rg) was employed to determine protein compactness. Before 100 ns, the Rg values of the gyrase C-backbone bound to allicin and ciprofloxacin were slightly deviated and subsequently obtained values between 21.5 and 22.0 Å at 200 ns (Figure 7 C1). On the contrary, ciprofloxacin-bound topoisomerase IV showed Rg values from 16.9 to 17.2 Å, while allicin-bound topoisomerase IV exhibited steady values between 14.4 and 14.6 Å (Figure 7 C2). Lower gyration (Rg) values suggested that the protein adopted a highly compact orientation when bound to a ligand. Moreover, the number of hydrogen bonds between the protein and the ligand indicated the complex's stability and strong interactions. Figure 7 D1 showed the greatest number of hydrogen bonds formed in the Gyrase-Ciprofloxacin complex at the beginning of the simulation while six hydrogen bonds formed in Topoisomerase IV-Ciprofloxacin during 70 to 80 ns (Figure 7 D2). Both ciprofloxacin-bound protein complexes had a greater number of hydrogen bonds than allicin-bound proteins throughout the 200 ns simulation. According to Figure 7 E1, the solvent-accessible surface area (SASA) values of Gyrase-Allicin and Gyrase-Ciprofloxacin were 13000–13500 Å2 and 14000–14500 Å2, respectively. The Topoisomerase IV-Allicin obtained 7000–7500 Å2 of SASA whereas the ciprofloxacin-bound protein obtained 8500–9000 Å2 (Figure 7 E2). When the value of SASA was decreased, the binding stability of the complex increased.
Molecular Mechanics Generalized Born Surface Area (MM-GBSA) Calculations
The binding free energy in the form of MM-GBSA was calculated for each of the combinations of the enzymes, Gyrase-Allicin, Gyrase-Ciprofloxacin, Topoisomerase IV -Allicin, and Topoisomerase IV -Ciprofloxacin using the MD simulation trajectory. Based on the findings, the protein-allicin complexes (Topoisomerase IV -Allicin, −85.9339 kcal/mol and Gyrase-Allicin, −160.518 kcal/mol) exhibited lower energies compared to protein-ciprofloxacin complexes (Topoisomerase IV -Ciprofloxacin, −50.2234 kcal/mol and Gyrase-Ciprofloxacin, −53.0945 kcal/mol). The energy of the complex was inversely proportional to the binding affinity.
Principal Component Analysis (PCA)
PCA was conducted to assess the structural variations in topoisomerase IV and gyrase bound to allicin and ciprofloxacin. The PCA scatter plots (Figure 8) illustrated the clustering and separation of principal components for each complex. In Topoisomerase IV -Allicin (Figure 8A), the principal component PC1 accounted for 41.81% of the variance, while PC2 explained 17.97%. The scatterplot showed two distinct clusters, indicating that allicin induced significant conformational changes in topoisomerase IV, maintaining stability with coordinated movements. In Topoisomerase IV -Ciprofloxacin (Figure 8B), PC1 contributed 36.58% of the variance and PC2 contributed 17.47%. This plot exhibited a more spread-out distribution, suggesting greater flexibility and less stable interaction in comparison to the allicin-bound complex. For Gyrase-Allicin (Figure 8C), PC1 accounted for 50.18% of the variance, with a clear clustering pattern that highlighted the stabilizing effect of allicin on gyrase. The tight clustering suggested that allicin promoted coordinated and stable interactions. In Gyrase-Ciprofloxacin (Figure 8D), PC1 captured 35.94% of the variance, showing a less defined clustering pattern, indicating more pronounced conformational changes and greater flexibility, similar to the Topoisomerase IV -Ciprofloxacin complex.
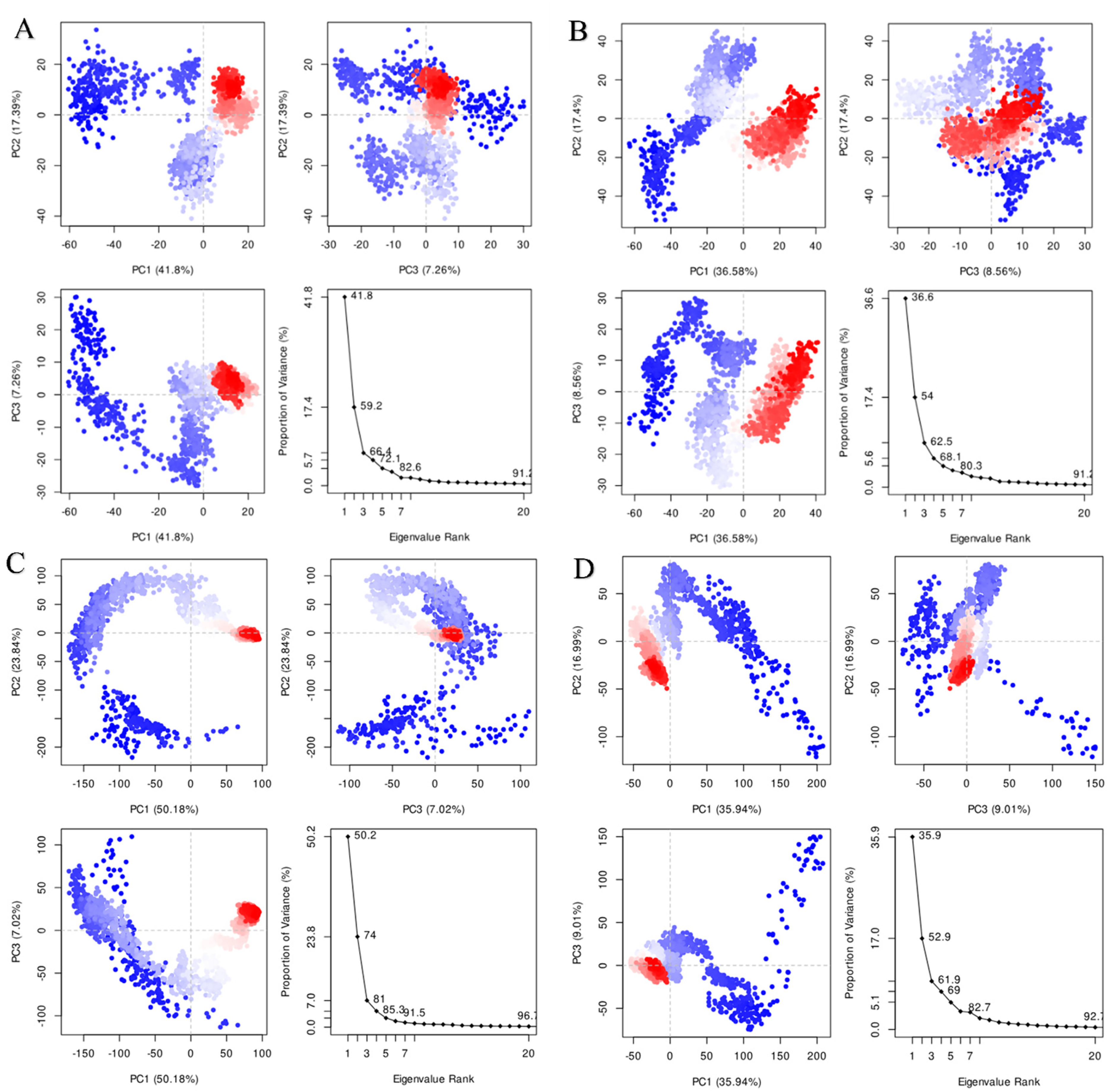
PCA of Topoisomerase IV and Gyrase in Complex With Allicin and Ciprofloxacin. (A) Topoisomerase IV-Allicin Complex. (B) Topoisomerase IV-Ciprofloxacin Complex. (C) Gyrase-Allicin Complex. (D) Gyrase-Ciprofloxacin Complex.
Dynamic Cross-Correlation Matrix (DCCM) Analysis
The DCCM analysis was performed to evaluate the interaction of topoisomerase IV with allicin and topoisomerase IV with ciprofloxacin, as well as gyrase with allicin and gyrase with ciprofloxacin. For the Topoisomerase IV-Allicin complex, the DCCM revealed regions with stable positive correlations, indicating coordinated movements between protein domains. A few areas showed weak anti-correlations, reflecting some flexibility within the structure (Figure 9A). Conversely, the DCCM for Topoisomerase IV-Ciprofloxacin displayed more pronounced anti-correlated motions, signifying that ciprofloxacin induced more disruptive interactions within the protein (Figure 9B).

DCCM Analysis of Topoisomerase IV and Gyrase in Complex With Allicin and Ciprofloxacin. (A) Topoisomerase IV-Allicin Complex Shows Positive Correlations. (B) Topoisomerase IV-Ciprofloxacin Complex Reveals More Anti-Correlated Motions. (C) Gyrase-Allicin Complex Exhibits Positive Correlations. (D) Gyrase-Ciprofloxacin Complex Displays Pronounced Anti-Correlations.
In the case of Gyrase-Allicin, the DCCM analysis showed several regions with positive correlations, implying that allicin induced stable, coordinated movements in gyrase (Figure 9C). On the other hand, the Gyrase-Ciprofloxacin complex exhibited more areas with anti-correlated motions, pointing to greater disruption and less coordinated movement between residues (Figure 9D). These contrasting results underscored the different ways allicin and ciprofloxacin influencing the dynamics of both topoisomerase IV and gyrase.
Discussion
Medicinal plants, such as garlic offer effective, affordable, and readily available alternatives for indigenous and traditional medicine, with direct antibacterial and antifungal properties.48,49 The antibacterial property of garlic is significantly contributed by allicin, a thiosulfinate found when garlic was injured. Based on the result of the in vitro experiment, allicin generated the largest inhibition zones on bacteria compared to the garlic extract and ciprofloxacin, indicating a better inhibitory effect against bacteria. Previous study stated that 50 µL of fresh garlic extract (6 g/mL) was more effective against S. aureus and E. coli than 30 µg/mL gentamicin, due to larger inhibition zones formed. 6 However, the agar diffusion assay was limited by several factors, including the concentration of antibacterial agents, their diffusion properties, and the tested bacteria's susceptibility. 50 Thus, the inhibition zone assay only served as the preliminary screening, demonstrating allicin had different degrees of inhibitory effect against all tested bacteria.Although allicin was tested at a higher concentration than ciprofloxacin, this was necessary due to its known instability and rapid degradation under assay conditions, as reported in previous studies. 51 In contrast, ciprofloxacin is a stable synthetic compound, effective at lower concentrations. Therefore, the higher initial dose of allicin was required to maintain a biologically active level throughout the experiment. The most popular methods in assessing the antibacterial activity of plant extracts or compounds are minimum inhibitory and bactericidal concentrations, 52 providing accurate quantitative measurements to compare with previous studies. In the present work, the concentrations of minimal inhibitory and bactericidal of allicin on the tested bacteria were lower than the garlic extract and ciprofloxacin, revealing greater antibacterial potency and effectiveness of allicin. Zainal et al (2021) stated that allicin had greater antimicrobial activity against S. aureus with 8 µg/mL of MIC and MBC, suggesting it is a potent therapeutic agent to treat Denture stomatitis. 53 The active compound in garlic, allicin demonstrates antimicrobial effects against various microorganisms.51,54–56 It inhibits RNA, DNA, and protein synthesis, disrupts lipid synthesis, and interferes with cell wall formation.57,58 Allicin offers natural alternatives to conventional antibiotics, with potential applications in treating bacterial infections. 59
By using the in silico methods, the target proteins and mechanism of allicin on bacteria can be further justified. In the present work, the consensus sequences for DNA gyrase and topoisomerase IV were predicted by aligning the protein sequences of all the test bacteria. The prediction of the consensus sequence assisted in designing more stable proteins with high biological activity. 60 The availability of several genome sequences has made it possible to compare the genomics of Pseudomonas aeruginosa in silico,61,62 revealing a conserved region shared among related species.63,64 The core genome, accounting for around 90% of P. aeruginosa's genome, demonstrates high conservation. 61 In molecular docking analyses, allicin was identified as a potential replacement for ciprofloxacin, a potent quinolone antibiotic. Ciprofloxacin demonstrated DNA breakage and cell-killing properties, including efficacy against a gyrase and topoisomerase IV.65–67 Ciprofloxacin stops bacterial growth by focusing on DNA replication by obstructing bacterial DNA gyrase and topoisomerase IV. Its selection as an antibiotic is supported by its well-established mechanism of action and effectiveness against various bacterial species, including P. aeruginosa. 65
In this study, the outcomes of ciprofloxacin and allicin were assessed by inhibiting DNA gyrase and topoisomerase IV. Based on the molecular docking studies, allicin and ciprofloxacin are bound efficiently against gyrase and topoisomerase IV. In previous work, ciprofloxacin analogs were docked to DNA gyrase's active site, which is the main target of antibiotics of the ciprofloxacin class. 68 Similarly, we have observed that ciprofloxacin has a significant affinity for gyrase and topoisomerase IV. On the other hand, allicin seemed to have better affinity depicted from docking studies over ciprofloxacin on topoisomerase IV. The binding affinity between gyrase and allicin was worse than ciprofloxacin but this result also gave some clues about the inhibitory mechanism of allicin. From the results of both target proteins, it was suggested that the allicin-bound protein exhibited greater stability due to greater interaction formed between the structures. By comparing all protein-ligand complexes, Topoisomerase IV-Allicin had the greatest binding affinity between the structure, indicating that topoisomerase IV is the main target of allicin compared to DNA gyrase and allicin had a better specificity with topoisomerase IV than ciprofloxacin.
Molecular dynamics simulation studies significantly explained from RMSD, radius of gyration, and SASA that allicin-bound gyrase and topoisomerase IV were more stable than ciprofloxacin-bound complexes. A previous study identified DNA gyrase as an allicin target and carefully examined it, utilizing in vitro and in silico techniques to give support. 17 RMSD studies from MD simulation trajectory explained a plausible justification regarding protein stability and system equilibration. Allicin-bound complex seemed to be higher stable than standard antibiotic ciprofloxacin complex. It is obviously observed from the results of MDS analysis that the stability of the Topoisomerase IV-Allicin complex was the best due to fewer fluctuations, compact structure, smallest solvent-accessible surface area and better structural convergence. Furthermore, the MM-GBSA studies confirmed a higher binding affinity of allicin compared to ciprofloxacin, as evidenced by more negative binding energies. 69 These findings suggest that allicin holds promise as a viable alternative to ciprofloxacin.
PCA was employed to investigate the collective motion of the protein-inhibitor complexes. 70 The PCA analysis revealed distinct dynamic behaviors for topoisomerase IV and gyrase when bound to allicin versus ciprofloxacin. The allicin-bound complexes displayed tighter clustering in the PCA plots, indicating that allicin promotes more stable and coordinated interactions within both topoisomerase IV and gyrase. This suggested that allicin may enhance the overall stability of these enzymes, potentially contributing to its inhibitory effects by reducing structural flexibility and promoting a more rigid, stable conformation. In contrast, the ciprofloxacin-bound complexes exhibited more dispersed clusters, signifying greater structural flexibility and less coordinated motion. The increased flexibility observed in these complexes suggests that ciprofloxacin may induce destabilizing effects, allowing for greater conformational variability and potentially reducing its efficacy as an inhibitor.
The DCCM analysis demonstrated that allicin induces greater coordination within both topoisomerase IV and gyrase compared to ciprofloxacin. For topoisomerase IV and gyrase, the stronger positive correlations observed in the allicin-bound complex suggest that allicin has a stabilizing effect on the enzyme's structure, potentially enhancing its inhibitory capabilities by promoting stability in critical functional domains. In contrast, ciprofloxacin induced more anti-correlated motions in both proteins, reflecting destabilization and reduced effectiveness as an inhibitor.71,72 These findings aligned with previous studies using DCCM to investigate ligand-induced stabilization and destabilization of proteins. Dos Santos Nascimento et al (2022) used DCCM to explore the stabilization of NLRP3 inhibitors, while Avti et al (2022) applied it to SARS-CoV-2 main protease inhibition. Both studies emphasize the importance of DCCM in revealing ligand-induced protein domain dynamics.71,72
Overall, allicin can act as an alternative drug to inhibit the DNA gyrase A and topoisomerase IV subunit A for bacterial infection treatment. This dual-targeting inhibition of the allicin can lower the probability of developing bacterial resistance. Moreover, allicin has greater specificity and exerts more inhibitory action on topoisomerase IV, compared to DNA gyrase. Once the allicin bound to topoisomerase IV, the structure of the Topoisomerase IV-Allicin complex was altered to promote the coordinated motion, forming a more rigid and stable structure. Greater interactions were formed within the complex to hold them more closely. The increased stability of the function domain enhances the inhibitory effect of allicin on topoisomerase IV. The binding of allicin with topoisomerase IV stabilizes the enzyme-DNA cleavage complex to increase the number of DNA break, inducing SOS response and mutagenesis of bacterial cells.73,74 The inhibition of allicin on topoisomerase IV also avoids the untangling of the daughter chromosomes resulting in halted or disastrous cell division. Finally, the cell death pathway of the bacteria is induced. 75
Allicin is an active compound that can be employed for a variety of medical purposes. Besides, it could be applied in combination with conventional antibiotics or antifungals to increase the successful rate of treatment when combating multidrug-resistant bacteria.6,55 Since allicin is widely used in therapeutic properties, its analogs could be synthesized as novel drug candidates.76,77 Regarding limitations, the significant protein residues interacting with allicin and contributing to protein stability and binding cannot be identified in the present work. Further analysis of these important residues should be conducted to explore their roles in the reaction and assist the rational design of the enzyme. Another limitation is that the molecular docking study omits the presence of the chelated divalent cation and explicit water molecules that significantly contribute to the binding between type II topoisomerases and ciprofloxacin. Future studies should include metal coordination and solvent effects to depict the binding mechanism more accurately.
Conclusion
In conclusion, allicin may interact more favorably with DNA topoisomerase IV subunit A than ciprofloxacin, based on in silico modeling and in vitro antibacterial assays. Bioinformatics tools provided insights into the potential molecular mechanism of allicin's action. Its predicted ability to promote stable, coordinated motions within topoisomerase IV supports its possible role as a novel antibacterial candidate. Further experimental validation is necessary to confirm these findings and assess therapeutic potential. Additionally, prediction of new protein structures from conserved genomic regions may assist in enhancing ligand-binding interactions for future drug development.
Supplemental Material
sj-docx-1-npx-10.1177_1934578X251368993 - Supplemental material for Novel Molecular Mechanism of Allicin-Mediated Inhibition of Bacterial Topoisomerase IV: A Combined Computational and Experimental Analysis
Supplemental material, sj-docx-1-npx-10.1177_1934578X251368993 for Novel Molecular Mechanism of Allicin-Mediated Inhibition of Bacterial Topoisomerase IV: A Combined Computational and Experimental Analysis by Ammar Khazaal Kadhim Almansoori, Raoof J. A. Maaroof, Ghadah Ali Al-Oudah, Nada Khazal K. Hindi, Lo Hui Yu, Haider Mahmod Jasim, Mohd Adnan and Mutaman Hussein Abdullah in Natural Product Communications
Footnotes
Acknowledgements
We would like to express our sincere gratitude to the Universiti Sains Malaysia, and Al-Mustaqbal University for providing the essential facilities for this study. We are also deeply appreciative of the College of Nursing, University of Babylon, for their unwavering support and provision of necessary resources, which were pivotal to the successful completion of this work.
Ethical Approval
Ethical approval is not applicable for this article.
Consent to Participate
Consent to participate is not applicable for this article.
Consent for Publication
The authors declare that they have no competing interests. Not applicable.
Author Contributions
Ammar Khazaal Kadhim Almansoori conceptualized the study. Methodology was developed by Ammar Khazaal Kadhim Almansoori and Nada Khazal K. Hindi. Formal analysis was performed by Ammar Khazaal Kadhim Almansoori, Lo Hui Yu, Nada Khazal K. Hindi, Mutaman Hussein Abdullah, and Raoof J. A. Maaroof. Investigation was conducted by Ammar Khazaal Kadhim Almansoori and Nada Khazal K. Hindi. The original draft was written by Ammar Khazaal Kadhim Almansoori, Lo Hui Yu, Nada Khazal K. Hindi, Mutaman Hussein Abdullah, and Raoof J. A. Maaroof, Mohd Adnan, Raoof J. A. Maaroof and Ghadah Ali Al-Oudah contributed to reviewing and editing. Bioinformatic analysis was conducted by Ammar Khazaal Kadhim Almansoori, Lo Hui Yu and Haider Mahmod Jasim. Nada Khazal K. Hindi and Ammar Khazaal Kadhim Almansoori supervised the research. Funding acquisition was handled by Ammar Khazaal Kadhim Almansoori.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subject.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Availability of Data and Materials
Applicable.
Supplemental Material
The online version contains Supplemental material available at…
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.