
Abstract
Objective
This study aimed to explore the potential targets and molecular mechanisms underlying the progression of acute liver injury, as well as to predict traditional Chinese medicines associated with key targets.
Methods
Differentially expressed genes (DEGs) associated with acute liver injury were first screened. Then immune infiltration analysis was performed using CIBERSORT algorithm, followed by Weighted Gene Co-Expression Network Analysis (WGCNA) analysis. Subsequently, Gene Ontology (GO) and Kyoto Encyclopedia of Genomes (KEGG) analyses of overlapping genes between DEG and WGCNA were performed using the DAVID database. The main active components of the predicted Chinese medicines were determined through network pharmacology analysis. In vitro experiment was performed to observe the effects of different concentrations of quercetin on the inflammatory response of LPS-induced J774A.1 macrophages.
Results
The results showed elevated M1 macrophages and activated dendritic cells in acute liver injury. A total of 40 DEGs associated with M1 macrophages were obtained, which were significantly enriched in four pathways, including the Toll-like receptor signaling pathway (P < .05). Furthermore, eight potential traditional Chinese medicines associated with Toll-like receptor signaling were identified, and network pharmacological analysis revealed quercetin as their main active ingredient. In vitro experimental results showed that quercetin reduced the mRNA levels of IL-6, TNF-α, IL-1β, and NF-κB as well as the protein levels of IL-6 and TNF-α in LPS-induced macrophages (P < .05). Additionally, quercetin significantly inhibited the release of NO (P < .05).
Conclusion
The present study provides further evidence that M1 macrophages potentially exert pro-inflammatory effects in acute liver injury via the toll-like receptor pathways. Notably, quercetin demonstrates inhibitory properties in this process, and its mechanism of action may be intricately linked to the TLR4-NF-κB signaling pathway.
Introduction
Acute liver injury is a disease characterized by rapid damage to hepatocytes and functional impairment within a short span.1,2 Its etiology is diverse, encompassing viral infections like hepatitis B and C, drug or chemical toxicity, alcohol abuse, autoimmune diseases, and metabolic disorders.3,4 It is vital to emphasize that a subset of patients with severe acute liver injury may progress to acute liver failure, characterized by a rapid decline in liver function due to massive hepatocyte death within a short period, resulting in irreversible damage. 5 Over the years, acute liver injury has emerged as a significant global health threat, owing to its diverse etiology, poor prognosis, and high mortality rates. Despite this, the precise mechanisms underlying acute liver injury remain unclear. 6 Therefore, identifying key molecules involved in this condition and implementing early intervention and appropriate treatments assume crucial significance in improving patient outcomes.
The process of rapid and massive hepatocyte death during liver failure is often accompanied by intense immune hyperactivity and inflammatory stress injury. Accumulating evidence has demonstrated the important role of inflammation in the pathogenesis of acute liver injury. It is a result of multiple interactions involving cell death molecules, immune cell-derived cytokines and chemokines, as well as damaged cell-released signals which orchestrate hepatic immune cell infiltration.7,8 Studies indicate that macrophages and lymphocytes play a role in amplifying the inflammatory response by releasing cytokines and chemokines. 9 The sustained inflammation disrupts normal liver function, impairs hepatocyte regeneration, and leads to the development of liver failure.10,11 However, the current research primarily focuses on the roles of specific immune cells or pathways in acute liver injury under certain etiological conditions. There is limited exploration from a big data perspective to comprehensively uncover the connections between immune cells and genes throughout the process of acute liver injury.
There is currently no effective therapy for acute liver injury other than liver transplantation.12–14 Consequently, there is a pressing need to investigate and discover novel drugs and approaches that can effectively alleviate liver injury. In recent years, traditional Chinese medicine has attracted increasing interest in the prevention and treatment of acute liver injury.15–17 Numerous studies have indicated traditional Chinese medicine can treat liver injury through multiple pathways and targets, such as suppressing immunoinflammation and anti-oxidative stress, and most of the herbal monomers in the treatment of liver injury are pre-protective to the liver by early administration.18–20 In particular, quercetin has become a promising candidate for the treatment of liver injury due to its powerful anti-inflammatory and antioxidative properties. Clinical studies have shown that many traditional Chinese medicines containing quercetin, such as Hedyotis diffusa, Ginkgo biloba, and Hypericum perforatum, have heat-clearing and detoxifying effects. It has been highly effective in treating inflammation-related diseases like Mycoplasma pneumonia, sepsis, and asthma.21,22 Compared to general anti-inflammatory drugs, quercetin, as a plant polyphenol, is widely present in various fruits and vegetables, and may be more effective in preventing diseases at an early stage.
In this study, we performed a comprehensive bioinformatics analysis to screen out critical genes associated with the immune response in acute liver injury. Subsequently, potential therapeutic traditional Chinese medicines targeting these genes were predicted. Cellular experiments were then conducted to validate their efficacy. The findings from this study offer novel insights and potential targets for the treatment of acute liver injury.
Materials and Methods
Data Collection
Gene microarray data related to acute liver injury in mice were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). A search and selection process were conducted using keywords such as “Acute liver injury”, “Acute liver failure”, “Fulminant hepatic failure”, “Acute hepatic necrosis”, and “Fulminant hepatic necrosis”. The inclusion criteria for the dataset required that the tissue samples originated from mice and included both a normal control group and an acute liver injury group induced by chemical agents. The final dataset comprised three microarray datasets: GSE167032, GSE167033, and GSE166488. For this study, mouse samples stimulated for one day were selected. The GSE167032 dataset included 5 samples of mice with acute liver injury induced by acetaminophen (APAP) and 5 samples of normal controls. The GSE167033 dataset consisted of 5 samples with acute liver injury induced by CCL4 and 5 samples of normal controls. The GSE166488 dataset contained 5 samples of mice with acute liver injury induced by LPS and 3 samples of normal controls. The microarray study type was mRNA, and the species analyzed were “Mus musculus”. Additionally, a validation dataset, GSE38941, was included, consisting of 10 human normal liver tissue samples and 17 human liver failure samples. All datasets underwent background correction and data normalization using the RMA algorithm.
Data Set Processing and DEGs Screening
The dataset was processed using R 4.0.5, and “limma” package was applied to identify differentially expressed genes (DEGs) with a threshold of adjusted P-value < .05 and |log2FC| > 1. This analysis successfully led to the identification of DEGs in the three datasets.
Immune Cell Infiltration and WGCNA Analysis
Immune Cell Infiltration Analysis
An Immune cell infiltration analysis was conducted to investigate the infiltration of immune cells in the studied samples. The original three control groups were merged into a new control group, while the LPS, APAP, and CCL4 groups were categorized as the acute liver injury group. The CIBERSORT algorithm was utilized to calculate the relative proportions of different immune cell types in each sample. The dataset “Mice” employed in this study, consists of 511 genes. 23 It facilitates the identification of 25 types of immune cells in mice and serves as an annotated gene set. The number of permutations was set to 1000, and the relative proportions of the two groups were compared using the Wilcoxon test. Subsequently, correlations between various immune cell types are calculated. The visualization of results is achieved using the “ggplot2” R package.
Weighted Gene Co-Expression Network Analysis
In order to investigate the relationship between gene expression and M1 macrophages, we utilized immune infiltration scoring and performed Weighted Gene Co-Expression Network Analysis (WGCNA). A soft threshold value of 8 was empirically selected as the highest value that did not exceed 0.85, as shown in Figure 1B. To classify genes with similar expression profiles into distinct modules, a hierarchical clustering dendrogram was constructed (minModuleSize = 30, mergeCutHeight = 0.25). Subsequently, module eigengenes (MEs) were calculated for each module, and the correlation between the MEs and the relative proportions of different macrophage types, as estimated above, was evaluated. With this approach, a set of hub genes associated with M1 macrophages was obtained in the context of acute liver injury. Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/) was employed to generate a Venn diagram visualizing the overlapping genes. This approach allowed for the identification of genes that exhibited both differential expression in liver injury and a strong association with M1 macrophages, providing valuable insights into the molecular mechanisms underlying liver injury.

Immune cell infiltration and WGCNA analysis. Immune cell infiltration analysis of three acute hepatic injury mice gene chips (A). Scale-free fitting index analysis of soft-thresholding powers (B). WGCNA screens out key genes related to M1 macrophages (C).
Enrichment Analysis
The obtained DEGs were uploaded to the DAVID platform (https://david.ncifcrf.gov/) for Gene Ontology (GO) functional analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. The background gene was defined as “Mus musculus”. For GO enrichment analysis, the DEGs were categorized into three main categories: biological process, molecular function, and cellular component. Enriched terms in each category were selected based on P-value < .05. Subsequently, the results were visualized using a bioinformatics platform (http://www.bioinformatics.com.cn/).
Network Pharmacology Analysis
Traditional Chinese Medicines Prediction of Key Genes
Selection of signaling pathways of interest from KEGG enrichment analysis, and importing genes enriched in the selected pathways into the Coremine Medical database (https://www.coremine.com/) for prediction of traditional Chinese medicines. The screening of potential therapeutic Chinese medicines was conducted using a significance level of P < .05.
Analysis of Active Components in Traditional Chinese Medicines
The efficacies and classification principles of the predicted traditional Chinese medicines were summarized. Then importing drugs into the TCMSP (https://old.tcmsp-e.com/tcmsp.php), and a screening condition of Oral bioavailability (OB) > 30% and drug-like properties (DL) > 0.18 was applied to identify their active ingredients and targets. These criteria are based on previous research.24,25 The target information was standardized using the Uniprot (https://www.uniprot.org/). Subsequently, the data were imported into Cytoscape 3.8.1 for visualization. The Cytohubba tool integrated within Cytoscape was employed for centrality analysis. Based on the degree values of the nodes, the highest-ranked active ingredient of the predicted drugs was obtained.
Cell Experimental Validation
Cell Culture
J774A.1 macrophage cells (Procell Life Science Technology Co., Ltd) were uniformly seeded in DMEM high-glucose medium supplemented with 15% FBS. The cells were cultured in a humidified incubator at 37 °C with 5% CO2, and the medium was changed every 2 days. Experiments were performed using cells in the logarithmic growth phase.
Cell Viability Assay by CCK-8
Cells were seeded in a 96-well plate at a density of 1 × 105 cells/mL, with a volume of 100 μL per well. The control group and quercetin (Sigma-Aldrich, USA, CAS: 117-39-5) treatment groups were set up at final concentrations of 0, 5, 10, 20, 40, 80, 160, 320, and 640 μmol/L. After 2 h of incubation, 1 μg/mL LPS (Sigma-Aldrich, USA, CAS: 93572-42-0) was added, and the cells were further cultured for 24 h. Prior to detection, 20 μL of CCK-8 solution (Zhuangmeng Biological Technology Co., Ltd, China, catalog number: ZP328) was added to each well and incubated for 1 h. The absorbance at 450 nm was measured using a microplate reader. The experiment was repeated at least three times.
Nitric Oxide (NO) Secretion Assay
Cells were seeded in a 6-well plate at a density of 3 × 105 cells/well and treated with quercetin at final concentrations of 10, 50, and 100 μmol/L for 2 h, followed by the addition of 1 μg/mL LPS for 24 h. The control group consisted of cells without any treatment, while the positive control group received 1 μg/mL LPS only. The culture supernatant was collected, and the NO content was measured according to the instructions provided in the assay kit (Jiancheng Bioengineering Institute, Nanjing, China, catalog number: A013-2-1). The experiment was repeated three times.
ELISA for Inflammatory Cytokine Secretion
In addition, the culture supernatant from each group of cells was collected, and the levels of IL-6 and TNF-α were determined using Enzyme linked immunosorbent assay (ELISA) kits IL-6 (BD Biosciences, USA, catalog number: 555240) and TNF-α (BD Biosciences, USA, catalog number: 555268) according to the manufacturer's instructions. The concentrations of the cytokines were calculated based on the standard curve.
qRT-PCR for mRNA Expression
Total RNA was extracted and quantified using an RNA extraction kit (Tianmo Biotech Co., Ltd, China, catalog number: TR205-5). Total RNA was quantified using a NanoDrop spectrophotometer (Thermo Scientific) to measure absorbance at 260 nm (A260) and assess the purity by the A260/A280 ratio. The extracted RNA was reverse transcribed into cDNA using a reverse transcription kit (Invitrogen, China, catalog number: K1691), and SYBR Green-based assays (ABclonal, China, catalog number: RK21204) were used to detect the mRNA expression levels of iNOS, IL-6, IL-β, TNF-α, and NF-κB. β-actin was used as an internal reference, and the relative expression levels of mRNA were calculated using the 2−ΔΔCt method. The RT-qPCR conditions were as follows: pre-denaturation at 95 °C for 3 min; 45 cycles of denaturation at 95 °C for 5 s, annealing at 60 °C for 34 s, The 20 μL reaction mixture was composed of 0.4 μL (10 μmol/L) forward primer, 0.4 μL (10 μmol/L) reverse primers, 2 μL cDNA, 7.2 μL H2O, and 10 μL 2× Universal SYBR Green Fast qPCR Mix. The target gene expression was calculated by normalizing it with the expression of β-actin using the 2−ΔΔCt method. 26 The specific primer sequences of the target genes are listed in Table 1.
Primer Sequence.

Statistical Analysis
Data analysis was conducted using SPSS 21.0 statistical software. The results are presented as mean ± standard deviation (SD). For comparisons among multiple groups, one-way analysis of variance (ANOVA) was employed, followed by the LSD-t test for post hoc pairwise comparisons. The Mann-Whitney U test was used for comparisons of specific gene expression levels between two groups. Statistical significance was defined as P < .05, indicating a significant difference.
Results
Differential Gene Analysis
After differential analysis, volcano plots were generated to visualize the DEGs. The GSE167032 dataset exhibited a total of 934 DEGs, with 571 genes downregulated and 363 genes upregulated (Figure 2A). In the GSE167033 dataset, there were 509 DEGs, with 371 genes downregulated and 138 genes upregulated (Figure 2B). The GSE166488 dataset displayed 123 DEGs, with 30 genes downregulated and 93 genes upregulated (Figure 2C). Additionally, heatmaps were constructed to illustrate the distribution patterns of DEGs from the three datasets between the experimental and control groups (Figure 2D-F).
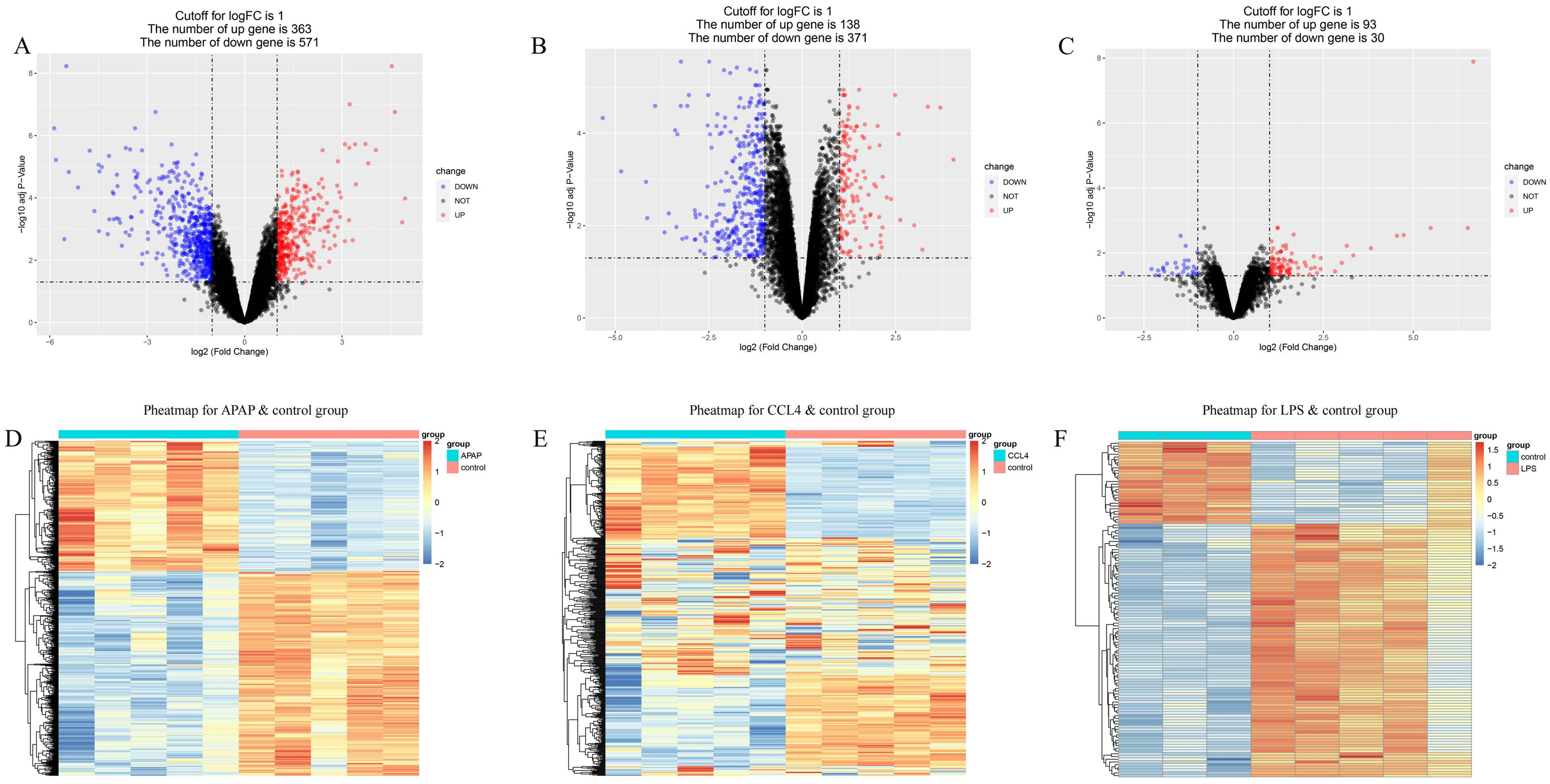
Heatmap and volcano plot of differentially expressed genes in three acute liver injury mice gene chips. Acute liver injury induced by APAP (A, D), CCL4 (B, E), and LPS (C, F).
Immune Cell Infiltration and WGCNA Analysis
The results of immune infiltration analysis revealed a significant increase in the proportions of M1 macrophages, activated dendritic cells, and a decrease in the relative proportions of mast cells, plasma cells, M0 macrophages, and helper T cells17 in acute liver injury compared to control tissues (Figure 1A). For the WGCNA analysis, fourteen modules were identified after merging highly correlated modules, as shown in Figure 1C. Among these, the gray-green module (r = 0.7, P = 3e-5) exhibited a positive correlation with M1 macrophage infiltration, and 131 genes within this module were further analyzed as M1 macrophage-associated genes. By intersecting the DEGs in acute liver injury with the genes in the weighted co-expression network, a total of 40 liver injury-related genes were obtained.
GO and KEGG Enrichment Analysis
To gain deeper insights into the various mechanisms of these genes, biological annotation was performed using the DAVID online database for the 40 common DEGs. GO functional enrichment analysis covered biological processes (BP), cellular components (CC), and molecular functions (MF). The results showed that the DEGs were primarily associated with biological processes such as acute-phase response, response to bacterium, response to stilbenoid, response to interferon-gamma, and regulation of immune system process. The molecular functions were related to extracellular region, extracellular space, high-density lipoprotein particle, and external side of plasma membrane. The cellular components were associated with small molecule binding, peptidase inhibitor activity, serine-type endopeptidase inhibitor activity, and heparin binding (Figure 3A). Furthermore, KEGG pathway enrichment analysis identified 4 signaling pathways, which were viral protein interaction with cytokine and cytokine receptor, Toll-like receptor signaling pathway, cytokine-cytokine receptor interaction, and cytosolic DNA-sensing pathway (Figure 3B).

Go biological enrichment and KEGG pathway enrichment. Bar plot showing the significantly enriched GO terms associated with the intersection genes (A). Bubble plot illustrating the significantly enriched KEGG pathways related to the intersection genes (B). The expression levels of CXCL9, CCL5, and IRF7 in the experimental and validation groups, compared to the control or normal group, HC: healthy control, *P ≤ .05, **P ≤ .01, ***P ≤ .001 (C).
Network Pharmacology Analysis
A total of three genes, CXCL9, CCL5, and IRF7 (Table 2), were enriched in the Toll-like receptor signaling pathway (Table 2). The results indicated that the expression levels of these genes were significantly higher in the liver failure groups compared to the control groups, and similar findings were observed in the validation dataset (Figure 3C). Subsequently, we performed a traditional Chinese medicine prediction for these genes using the Coremine Medical database. A significance level of P < .05 was considered statistically significant, and a total of 42 different Chinese medicines were obtained (Table 3).
KEGG Analysis of 40 Hub mRNAs.

Traditional Chinese Medicines Prediction Results of Key Genes.
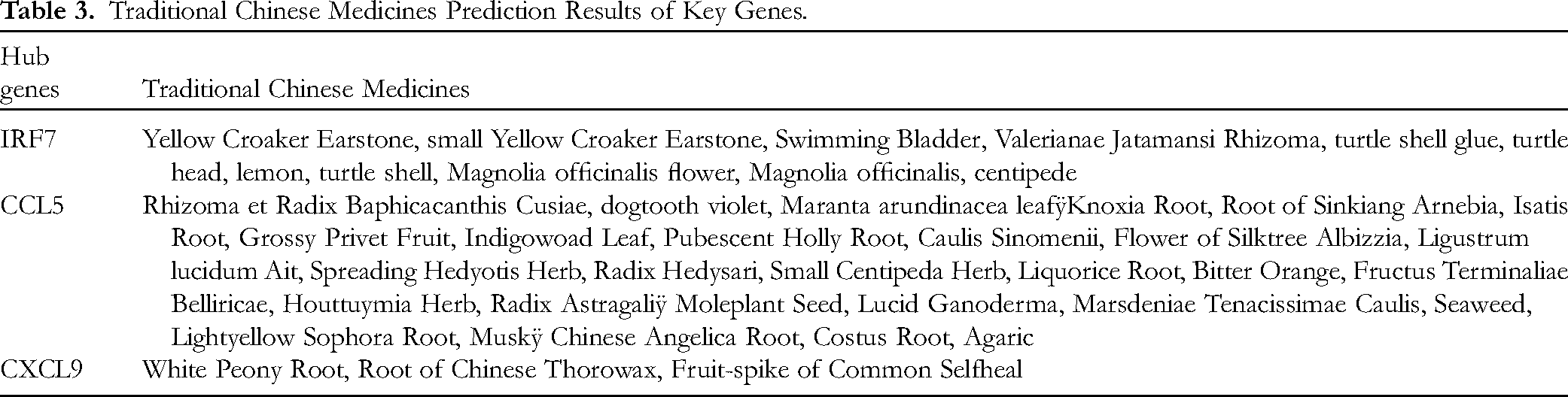
In traditional Chinese medicine, the pathogenesis of liver failure is believed to involve the “accumulation of heat-toxin and stasis of blood”. Therefore, candidate drugs were selected with the efficacy of clearing heat and detoxification, promoting blood circulation and removing blood stasis, and belonging to the liver meridian. The identified drugs were then imported into the TCMSP database to retrieve their chemical constituents. Selection criteria for screening were set as OB > 30% and DL > 0.18. As a result, active ingredients and their corresponding target proteins were obtained for eight candidate drugs, including Root of Sinkiang Arnebia, dogtooth violet, Houttuymia Herb, Fruit-spike of Common Selfheal, Lightyellow Sophora Root, Magnolia officinalis, Spreading Hedyotis Herb, Indigowoad Leaf.
A “Drug-Active Ingredient-Target” network diagram (Figure 4) was constructed using Cytoscape 3.8.1. The network diagram consisted of 287 nodes and 897 edges. The degree centrality of nodes was calculated using the cytohubba tool integrated in Cytoscape software. The results showed that quercetin (degree = 146), kaempferol (degree = 59), luteolin (degree = 56), beta-sitosterol (degree = 39), Indirubin (degree = 32), and Stigmasterol (degree = 32) were the top six active compounds with high degree values. These compounds may serve as core components for the treatment of liver failure, potentially exerting their effects through interactions with multiple target proteins.
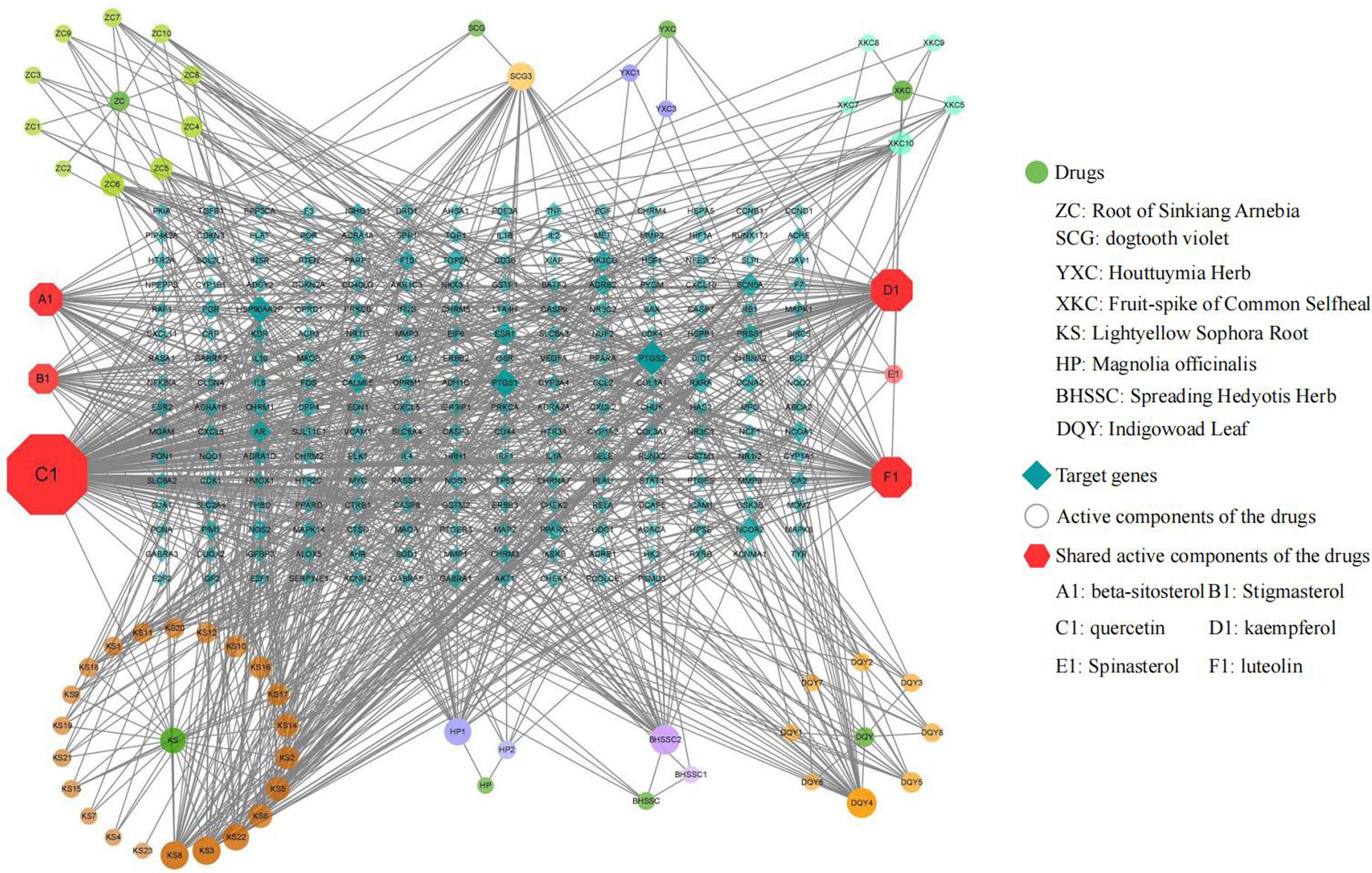
Network diagram of drug-active ingredient-target interactions. Red octagonal nodes represent shared active components of the drugs, blue-green diamond nodes represent target genes, and dark green circular nodes represent the drugs. Circular nodes branching out from each drug represent unique active components of that drug. The color and size of the nodes in the network graph correspond to their relative importance, with darker and larger nodes indicating greater significance.
Experimental Results
Effects of Quercetin on Macrophage Viability
Compared to the control group, the viability of macrophages gradually increased with the increasing concentration of quercetin, reaching a peak at 10 μmol/L after 2 h of treatment, and then gradually decreasing. Quercetin at concentrations of 160, 320, and 640 μmol/L significantly reduced macrophage viability (P < .05) (Figure 5A). After stimulating J774A.1 macrophages with 1 μg/mL LPS for 24 h, the effects of quercetin on macrophage viability were observed in an inflammatory environment. Compared to the LPS stimulation group, quercetin at concentrations of 5 and 10 μmol/L enhanced the viability of LPS-induced macrophage, while quercetin at concentrations of 80, 160, 320, and 640 μmol/L significantly reduced cell viability (P < .05) (Figure 5B).

Effects of quercetin on macrophage viability and LPS-induced macrophage viability. “*” indicates comparison with the control group, where *P ≤ .05, **P ≤ .01, ***P ≤ .001. “#” indicates comparison with the LPS group, where #P ≤ .05, ##P ≤ .01, ###P ≤ .001.
Effects of Quercetin on LPS-Induced Macrophage Inflammatory Factors
Changes in cytokine expression were detected using qRT-PCR. The results showed that compared to the LPS stimulation group, the mRNA levels of IL-1β and TNF-α were significantly decreased after treatment with 50 and 100 μmol/L of quercetin, and the mRNA levels of IL-6 was reduced after treatment with 100 μmol/L of quercetin, with statistical significance (P < .05) (Figure 6A). The levels of cytokines secreted by macrophages in the cell culture supernatant were measured using ELISA. The results showed that compared to the LPS stimulation group, the levels of IL-6 and TNF-α secreted by macrophages in the quercetin treatment group were significantly decreased, with statistical significance (P < .05) (Figure 6B).

The effect of quercetin on pro-inflammatory cytokine expression. Compared to the LPS-stimulated group, *P ≤ .05, **P ≤ .01, ***P ≤ .001.
Effects of Quercetin on NO Release, iNOS and NF-κB mRNA Levels
Compared to the LPS stimulation group, treatment with 50 and 100 μmol/L of quercetin significantly inhibited the release of NO in macrophages, with statistical significance (P < .05) (Figure 7A). Further analysis revealed that treatment with 50 and 100 μmol/L of quercetin significantly downregulated the expression of iNOS and NF-κB mRNA levels in LPS-induced macrophages, with statistical significance (P < .05) (Figure 7B and C).
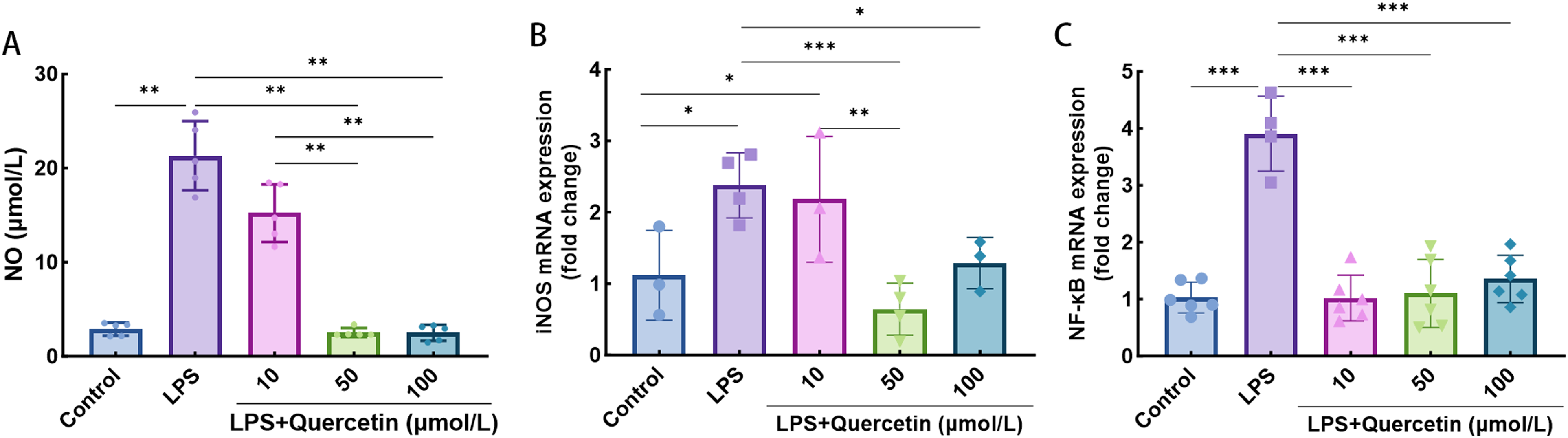
The effect of quercetin on NO release, iNOS mRNA levels and NF-κB mRNA levels in LPS-stimulated macrophage. Compared to the LPS-stimulated group, *P ≤ .05, **P ≤ .01, ***P ≤ .001.
Discussion
The precise etiology of acute liver injury can be multifactorial. Regardless of the initial trigger, a common hallmark of acute liver injury is the profound dysregulation of inflammatory processes within the liver, which further exacerbates the disease progression and contributes to organ damage.14,27,28 In this study, we conducted a comprehensive search of databases and integrated three datasets on acute liver injury, utilizing a bioinformatics approach to unravel the underlying molecular mechanisms associated with acute liver injury, with a specific focus on the interplay between liver injury and inflammation.
Macrophages, as important components of the immune microenvironment, play a crucial role in various diseases.29,30 The immune cell infiltration results obtained in this study revealed a significant increase in M1 macrophages in the acute liver injury group compared to the control group. Numerous studies have demonstrated the vital role of M1 macrophages in acute inflammation-related diseases. For instance, Wu et al 31 reported that M1 macrophages dominate during the pro-inflammatory phase of AP in a mouse model of acute pancreatitis induced by blind saccharin. Moreover, a study performed transcriptome sequencing after constructing an LPS/D-GalN-induced acute liver failure mouse model and found that M1 macrophages were higher in the liver failure group. 32 Taken together, the evidence strongly suggests that macrophages, as the most abundant immune cells in the liver,33–35 play a crucial role in the occurrence and progression of acute liver injury.36–38 Targeting macrophages may be a prospective therapeutic approach to modulate inflammation and facilitate liver regeneration. Additionally, there was a notable elevation in conventional dendritic cells in the experimental group. This finding is consistent with previous findings showing that conventional dendritic cells produced IL-10 and TGF-β through TLR9 activation during acute liver injury. 39 In short, immune infiltration results suggest a pronounced immune response associated with acute liver injury, characterized by an enhanced presence of pro-inflammatory immune cells and a reduction in regulatory immune cell populations.
To elucidate the pathway through which M1 macrophages exert their effects, we conducted a comprehensive pathway enrichment analysis. The results highlighted two predominant pathways, namely the Toll-like receptor signaling pathway and the cytokine signaling pathway. Both pathways are well-known for their crucial role in the immune system and close association with the inflammatory process. Toll-like receptors (TLRs) are recognized as a conserved set of pattern recognition receptors that initiate signaling cascades leading to immune responses upon activation by various pathogen-associated molecular patterns. 40 Our enrichment analysis further underscored the significance of TLRs in the context of acute liver injury.
Quercetin, a member of the flavonoid compound family, is abundantly present in traditional Chinese medicines like Bupleurum, Fritillaria, and Mulberry Leaf. 41 In this study, we focused on quercetin's impact on LPS-treated macrophages to investigate its anti-inflammatory activities. A pattern-recognition receptor for LPS is TLR4. 42 TLR4 recognizes LPS and activates the NF-κB signaling pathway, promoting the secretion of TNF-α, IL-6, IL-1β. Our experimental results showed that quercetin significantly reduced the mRNA and protein levels of these cytokines, indicating its strong anti-inflammatory effects. Regarding the mechanism by which quercetin exerts its anti-inflammatory effects through TLR4, Previous studies have revealed that quercetin inhibits signaling pathways such as NF-κB and MAPK, reducing the production of pro-inflammatory cytokines.20,43,44 Specifically, quercetin may compete with LPS for binding to TLR4, preventing LPS from activating TLR4, and thus inhibiting the subsequent NF-κB signaling pathway. Research has shown that quercetin can inhibit the expression of TLR4 and regulate its downstream signaling pathways, such as MyD88, IRAK4, and NF-κB, thereby reducing the inflammatory response.45,46 Additionally, quercetin may also alter the conformation of TLR4 or modulate its expression level, 47 further suppressing the immune response mediated by TLR4. Although direct experimental evidence demonstrating quercetin's binding to TLR4 is still lacking, the existing literature supports the idea that quercetin interferes with the TLR4 signaling pathway through the mechanisms mentioned above. Therefore, we hypothesize that quercetin's anti-inflammatory effects may be achieved by competing with LPS for TLR4 binding or by regulating TLR4 activity.
Interestingly, quercetin appears to have a dose-dependent bidirectional effect on macrophage proliferation. At lower concentrations (≤20 μmol/L), quercetin promotes macrophage proliferation, while higher concentrations of quercetin (>20 μmol/L) inhibit macrophage proliferation. This effect was also observed in LPS-treated macrophages. These findings align with a study by Xu et al, who obtained similar results when treating breast cancer cells with quercetin. 48 These results indicate that the effects of quercetin on macrophages may be dose-dependent. Therefore, the dosage of quercetin should be carefully considered in future applications to optimize clinical efficacy. However, quercetin's absorption and metabolism affect its bioavailability, and the metabolic transformation alters its biological activities.41,49 This may also be one of the reasons for the varying efficacy observed in other studies. Therefore, although our study provides some potential for drug development, the dose-dependent effects of quercetin, along with its absorption, distribution, metabolism, and excretion characteristics, should be considered to achieve optimal therapeutic outcomes.
This study presents a comprehensive investigation into the underlying molecular mechanisms contributing to the occurrence and development of acute liver injury, employing multiple approaches. In contrast to previous research, we not only explore immune cells and signaling pathways involved in inflammation during acute liver injury using bioinformatics methods but also investigate the potential of traditional Chinese medicines in treating acute liver injury through network pharmacology, with preliminary validation through cell experiments. Nevertheless, it is essential to acknowledge the limitations of this study. On the one hand, although many studies have shown hepatoprotective effects of quercetin in animal models, we need to establish an in vivo model to further validate our results. On the other hand, since we employed a mouse microarray for molecular level analysis and validated the findings using mouse cells, further validation is necessary to ascertain the applicability of the results to human systems. Additionally, protein-level validation of the identified biomarkers and pathways was not performed in this study. In summary, although quercetin demonstrates considerable potential in inhibiting inflammation, further in vivo studies are warranted to explore its anti-inflammatory mechanisms in greater depth. Additionally, larger-scale animal experiments are necessary to validate its efficacy and safety.
Conclusion
In conclusion, this study reveals the critical role of M1 macrophages and toll-like receptor pathways in the development of acute liver injury in a big data perspective. Furthermore, we conducted additional predictions and network pharmacology analyses of traditional Chinese medicines, revealing that quercetin is the main active ingredient for the Chinese medicine to function. Quercetin appears to exhibit a certain inhibitory effect on inflammation associated with acute liver injury. Subsequent experiment validates the significant potential of quercetin in treating inflammation associated with acute liver injury, as it can suppress macrophage inflammation mediated by LPS. Preliminary assessments suggest that the underlying mechanism may involve the regulation of the TLR4-NF-κB signaling pathway by quercetin. In brief, this study further informs the treatment of acute liver injury, targeting macrophages may be a prospective therapeutic approach to modulate hepatic inflammation, and quercetin is a pharmacological ingredient that deserves further investigation for clinical treatment. Future research should focus on optimizing quercetin's pharmacokinetics, conducting larger-scale clinical trials, and exploring its combination with other therapeutic agents to enhance its efficacy. Additionally, understanding the precise molecular mechanisms underlying quercetin's effects on macrophages and TLR4 signaling could lead to the development of more targeted therapeutic strategies.
Footnotes
Acknowledgements
Not applicable.
Abbreviations
GEO, Gene Expression Omnibus; DEGs, Differentially expressed genes; WGCNA, Weighted gene co-expression network analysis; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; OB, Oral bioavailability; DL, drug-like properties; CCK-8, Cell Counting Kit-8; ELISA, Enzyme linked immunosorbent assay; qRT-PCR, real-time quantitative PCR.
Author Contributions
We declare that all authors made significant contributions to this manuscript. XL and HY designed the study. XL, SK, JL, and XF collected and analyzed the data. XF, YL, XK, and SL performed the experiments. XL and SK wrote the manuscript. YW and JL advised on the experiments. HY provided critiques and revised the manuscript. All authors revised the manuscript and approved the final version.
Consent for Publication
Not applicable.
Data Availability
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate
Not applicable.
Funding
This work was supported by the Hebei Traditional Chinese Medicine Scientific Research Project (grant No.2021226) and the Key Research and Development Program Projects of Hebei Provincial Department of Science and Technology (grant No.223777156D).