
Abstract
Background
Esherichia coli is a Gram bacteria and a normal flora of the large intestines of human and other warm-blooded animals. 1 Unhygienic practices have made its isolation to be common among edible foods and vegetables. 2 Most strains are harmless, but some strains acquire bacteriophage or plasmid DNA-encoding enterotoxins or invasion factors that make them pathogenic or virulent. 3 These virulent strains are responsible for diarrheal infections worldwide, and other infections such as neonatal meningitis, septicemia, and urinary tract infections (UTIs). The most common pathogenic strains of E. coli are enterotoxigenic E. coli (ETEC), enteroinvasive E. coli (EIEC), Shiga toxin-producing E. coli (STEC), enteroaggregative E. coli (EAEC), EIEC, enteropathogenic E. coli (EPIC), and diffusely adherent E. coli (DAEC). Their virulence is attributed to the virulent factors they produce, which include colonization and adhesion factors, the production of toxins, and effectors.3,4
Added to these virulent factors is the ability of this pathogen to develop antimicrobial resistance to commonly used antibiotics in its management, 5 making it to be enlisted as one of the priority pathogens by the World Health Organization (WHO). 4 There are numerous reports of multi-drug resistance among E. coli isolates from various samples.1,6‐8 The incidence of infections caused by multidrug-resistant strains of E. coli is on the rise. 7 Added to this public health challenge is the fact that they have an outstanding ability to act as donors or recipients of resistant genes. 7 Commonly used antibiotics that are used to manage infections caused by species of the Enterobacteriaceae, such as extended-spectrum penicillins, cephalosporins, monobactams, carbapenems, fluoroquinolones (ciprofloxacin), and aminoglycosides (gentamicin), are not spared the multidrug resistance feat of E. coli.9–12 Multidrug-resistant E. coli is on the WHO's list of priority and critical pathogens for which new antibiotics are urgently needed.4,12 Members of the critical pathogens list, especially the multidrug pathogens, have been implicated in various hospital and nursing home infections.4,12
Developing new antibiotics is a time-consuming and financially demanding process. 13 To save time and money, researchers have turned to several strategies. In one study, researchers utilized machine learning, bioinformatics and explainable artificial intelligence to screen numerous compounds. 14 Yet others have turned to screening bioactive compounds from medicinal plants using in silico approaches.15,16 One of such plant is Tetrapluera tetraptera, locally called Uyayak in Cross River State, Southern Nigeria. The extracts of the fruit, leaves, stem, root, and essential oils of T. tetraptera have been shown to contain bioactive compounds that are responsible for its antimicrobial (bacteria and fungi) activities.17–21 Lin et al 17 revealed the mechanism of action of the root extract of T. tetraptera to be via eliciting severe damage to the integrity of the E. coli cell and its membrane permeability. These findings, put together, indicate that the various extracts and their bioactive compounds have excellent antimicrobial activities that need further evaluations. To the best of our knowledge, no study has utilized an in silico approach to evaluate the antimicrobial activity of the T. tetraptera bioactive compounds against any target protein of E. coli. Hence, the aim of this study was to conduct an in silico assessment (molecular docking) of the bioactive compounds revealed by Gas chromatography coupled to mass spectrophotometry (GC-MS) in addition to the evaluation of the drug-likeness via the prediction of their pharmacokinetic properties.
Methods
Collection of Fruit, Fruit Salad and T. tetraptera Samples
T. tetraptera (Uyayak) pods (Figure 1) were purchased from Marian and Watt Markets, respectively, in August 2022 and also stored in a Ziploc bag where they were transported without ice and within one hour of collection.

Whole fruit pod (left) of the T. tetraptera sample used in the study and the 3D crystal structure of a binary complex of E. coli dihydropteroate synthase (right) retrieved from protein data bank.
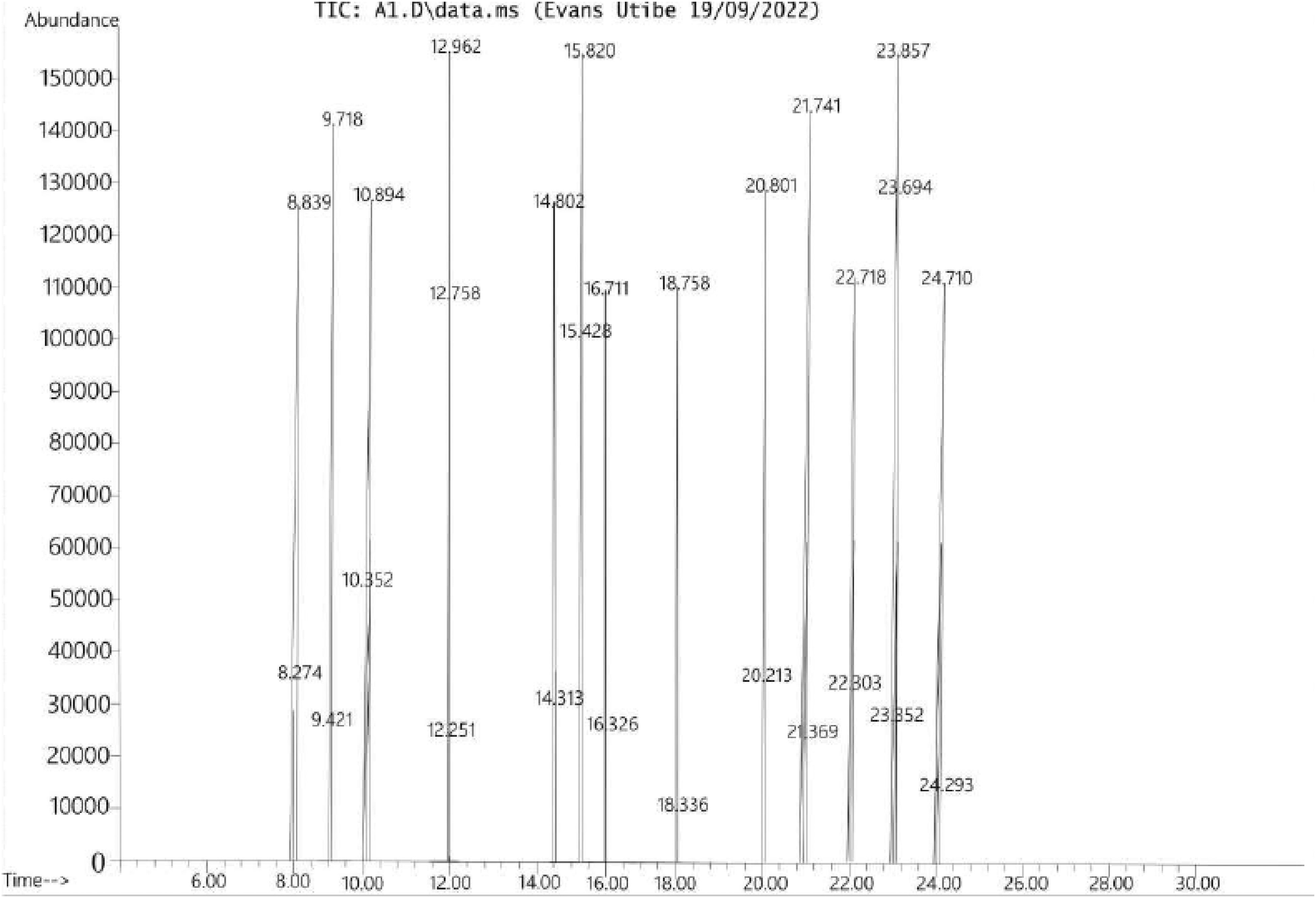
GC-MS chromatogram showing the peaks of the various bioactive compounds obtained from T. tetraptera pod in this study.
GC-MS Analysis of T. tetraptera
This was done as reported previously.22–25 Exactly 10 g of the milled T. tetraptera pod was added to 20 mL of methanol, and the resulting mixture placed on an orbital shaker for 15 min. The mixture was allowed to stand for another 15 min after shaking. The mixture was then filtered, and the filtrate concentrated to 5 mL using a rotatory evaporator. From the concentrate, bioactive compounds were screened using an Agilent 5890N gas chromatograph fitted with an autosampler connected to an Agilent mass spectrophotometric detector. All the operating conditions were as previously reported. 26 The bioactive compounds in the sample were identified and interpreted using the database of the National Institute of Standards and Techniques (NIST). 26
Prediction of ADMET Properties of the Ligands and Target Prediction
Bioactive compounds were screened for their absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties using the pkCSM prediction tool. The pkCSM tool is a well-established tool for the prediction of ADMET properties of lead drug compounds. 27 First, the ligands were converted into their respective SMILE strings at PubChem (https://pubchem.ncbi.nlm.nih.gov/). The strings were then used individually to predict the ADMET properties of the various ligands, together with their molecular descriptors. Using the online SWISSADME tool, potential targets were predicted and the top ten targets displayed. 28
Lipinski Rule of Five (ROF) Screening
The ligands (bioactive) compounds obtained from the GC-MS analysis were further screened for Lipinski's rule of five (ROF) using the SWISSADME tool. The SWISSADME is a free-web tool used in the prediction of lead compounds or small molecules pharmacokinetics, drug-likeness, and medicinal chemistry friendliness. 26 Following Lipinski's ROF screening, ligands that meet the ROF were further prepared for molecular docking.
Ligand Preparation for Docking
The screened ligands were prepared for molecular docking using various tools. First, the structures of the ligands were retrieved from their unique SMILES strings using the Chem3d 15.1 tool. Following retrieval of their structures, the energies of the ligands were minimized and then saved in pdb files. The minimized ligands were further prepared using AutoDock 4.2 and saved in pdbqt files individually. 29
Preparation of Proteins for Molecular Docking
From the Research Collaboratory for Structural Bioinformatics (RCSC) protein database (https://www.rcsb.org/), the dihydropteroate synthase of E. coli was retrieved. The details of the protein as obtained from the MCULE tool and RCSC PDB were 1AJ2, DHPS_ECOLI, P0AC13, and 83333 for the PDB ID, Uniprot name, Uniprot accession ID, and Uniprot taxonomic ID, respectively. As a protein complexed with native ligands (n = 2), the binding or active site of the protein was retrieved using these native ligands as a guide. The definition of the binding site was done using Biovia Discovery Studio Client 21. The binding site was defined to be x = 41.244328, y = 4.789960, and z = 8.014888, with a radius of 15.434639. The protein was further prepared by removing water molecules, and the resulting protein structure saved in pdb format. The protein in pdb was converted into a pdbqt file using AutoDock 4.2 for docking. 29
Molecular Docking and Visualization
Molecular docking was performed by AutoDock Vina version 4.2. 29 First, the various AutoDock files were retrieved and configured accordingly to align with the various prepared ligands and protein. Using the docking command lines, docking was performed, and the resulting docking modes were split for each ligand. The various interactions in the various binding modes were then visualized in 2D using the Biovia Discovery Studio Client 21 software to display the amino acids, types of bonding, and bond lengths.
Results
Figure 2 shows the GC-MS chromatogram (abundance vs retention time) of the bioactive compounds from T. tetraptera. The bioactive compounds are represented by the various peaks. The peaks were 28, representing a total of 28 bioactive compounds. Table 1 shows the retention time (min) of the 28 bioactive compounds, their molecular weights, peak areas (%), and development (min). The retention time ranged from 8.274 to 24.710 min for the compounds, methone and beta-caryophyllene. On the other hand, the molecular weights of the various compounds ranged from 16.04 to 478.7 g/mol for methone to carpaine, respectively. Peak area shows that lupeol was the most abundant with a peak area of 21.71, while eugenol had the least peak area with a value of 1.13. This implies that lupeol was the concentrated biaoctive compound while the least was eugenol. Classification of the bioactive compounds indicates that the compounds were diverse with monoterpenes (n = 8) being the most abundant in term of number and this was followed by sesquiterpene (n = 3). Other classes of compounds were triterpene (n = 1), sesquiterpene alcohol (n = 1), monoterpenes alcohol (n = 1), monoterpenes ketone (n = 1), dihydrazine (n = 1), hydroxy fatty acid (n = 1), triterpenoid (n = 2), fatty alcohol (n = 1), alcohol (n = 1), phytosterol (n = 1) and phenylpropanoid (n = 1).
Names, Retention Times, Molecular Weights, Peak Area and Development Times of the Various Bioactive Compounds in Our Studied T. tetraptera Pod Revealed by GC-MS.

Key = - no class.
Table 2 shows the predicted ADMET properties for ligands that had no violation of Lipinski ROF. Adsorption was evaluated using water solubility, intestinal absorption, skin permeability, P-glycoprotein substrate, and P-glycoprotein I and II inhibitors. The water solubility values of the study bioactive compounds ranged from −2.25 to 5.477, while those of trimethoprim was −2.744, which was within the range of our study ligands. Intestinal absorption for the various ligands ranged from 91.485 to 95.257 and was higher than that of trimethoprim, which was 71.787. Skin permeability for the ligands ranged from −1.063 to −2.715 while that of trimethoprim was −2.735. A total of 11 ligands were not substrates for P-glycoprotein while ligands II and X as well as trimethoprim were substrates. All the ligands, as well as trimethoprim, were not inhibitors of P-glycoprotein I and II. Distribution was evaluated using VDss (human), fraction unbound (human), BBB, and CNS permeabilities. From the predicted result presented in Table 2, the VDss values ranged from −0.574 to 0.812 for the ligands and 0.954 for the control drug. Fraction unbound values ranged from 0.0104 to 0.514 for the ligands and 0.554 for trimethoprim. For BBB and CNS permeabilities, the values for the ligands ranged from 0.084 to 0.792 and −2.977 to −1.763, respectively, while for trimethoprim, the respective values were −1.225 and −3.273 for BBB and CNS permeabilities. The predicted parameters for metabolism were CYP2D6 and CYP3A4 substrates and CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 inhibitors. From the predicted result presented in Table 2, none of the ligands nor trimethoprim were substrates for CYP2D6 while for CYP3A4, 11 ligands were not substrates, but ligands 10, 13, and trimethoprim were substrates. All the ligands as well as the trimethoprim were not inhibitors of CYP2C19, CYP2C9, CYP2D6, and CYP3A4; however, ligand VIII was predicted to inhibit CYP1A2. The predicted excretion parameter showed that total clearance ranged from 0.191 to 1.86 for the ligands, while it was 0.98 for trimethoprim. All the ligands and trimethoprim were not substrates for renal OCT2. Ames's toxicity profiling of the ligands and trimethoprim showed that all the ligands, apart from VIII and trimethoprim, could be carcinogenic. On the other hand, all the ligands did not indicate hepatoxicity concerns, while trimethoprim was predicted to be hepatotoxic. Furthermore, all the ligands, as well as trimethoprim, were not inhibitors of hERG I and II.
Predicted ADMET Properties for Ligands That Meet Lipinski's Rule of Five.

Key: I = Methone, II = Lemonene, III = Octanol, IV = Menthol, V = Dihydrocarvone, VI = Piperitone, VII = Terpinene-4-ol, VIII = Eugenol, IX = 15-Hydroxypentadecanoic acid, X = Carpaine, XI = T-Cadinol, XII = Alpha-ocimene and XIII = hexadecanoic acid.
Figure 3A and B show the summary of the various predicted targets for the various ligands and trimethoprim. The result showed various targets that ranged from 6 to 10 targets for the various bioactive compounds, while for trimethoprim, the number of targets was 7. Figure 4 shows a summary of the top predicted targets for the various ligands and trimethoprim. The top targets were family A G protein-coupled receptors, the fatty acid binding family, oxidoreductase, enzyme, and lyase for the ligands, while for trimethoprim, it was kinase. The most frequent target was the nuclear receptor, which was the most frequent target in six of the 13 bioactive compounds.

(A) A summary of target prediction results for ligands I to VI. Key: I = Methone, II = Lemonene, III = Octanol, IV = Menthol, V = Dihydrocarvone, VI = Piperitone. (B) A summary of target prediction results for ligands VII to XII and trimethoprim. Key: VII = Terpinene-4-ol, VIII = Eugenol, IX = 15-hydroxypentadecanoic acid, X = Carpaine, XI = T-cadinol, XII = Alpha-ocimene and XIII = Hexadecanoic acid and XIV = Trimethoprim.

A bar chart presentations of the top targets for each of the ligand and control. Key: M1 to M14 = Methone, Lemonene, Octanol, Menthol, Dihydrocarvone, Piperitone, Terpinene-4-ol, Eugenol, 15-hydroxypentadecanoic acid, Carpaine, T-cadinol, Alpha-ocimene, Hexadecanoic acid, and Trimethoprim, respectively. FAG = Family A G protein coupled receptor, FABF = fatty acid binding family, OX = Oxidoreductase and NR = Nuclear receptor.
Figure 5 shows the various interactions between the various ligands and trimethoprim against the dihydropteroate synthase of E. coli. The various docking poses showed the various bond lengths and amino acid residues that were involved in the interactions between the ligands and the enzymes. Table 3 shows the various amino acids involved in the interactions and their docking scores. The docking scores of the various ligands ranged from −4.0 to −5.3 kcal/mol, and this was lower than that of trimethoprim, which returned a docking score of −6.5 kcal/mol. However, one of the ligands, carpaine, returned a positive docking score of +44.3 kcal/mol, indicating unfavorable docking. The interacting amino acid residues varied among the various amino acids (Figure 5). Furthermore, the most common amino acid residue among the bioactive compounds was isoleucine 117, followed by threonine 62, as both residues were found to be involved in eight and six complexes, respectively. The amino acids also varied in terms of number. Trimethoprim complexed with 9 amino acids, and this was the highest, while the bioactive compounds or test ligands complexed with 2-7 amino acid residues. However, carpaine complexed with a total of 8 amino acids.

Docking outputs for the various ligands and trimethoprim visualized in 2-D using Biovia Discovery Studio 21. Key: I = Methone, II = Lemonene, III = Octanol, IV = Menthol, V = Dihydrocarvone, VI = Piperitone, VII = terpinene-4-ol, VIII = eugenol, IX = 15-hydroxypentadecanoic acid, X = carpaine, XI = T-cadinol, XII = alpha-ocimene and XIII = hexadecanoic acid and XIV = Trimethoprim.
Amino Residues Involved in Binding of the Various Bioactive Compounds Against Dihydropteroate Synthase and Their Docking Scores.

Key: I = Methone, II = Lemonene, III = Octanol, IV = Menthol, V = Dihydrocarvone, VI = Piperitone, VII = Terpinene-4-ol, VIII = Eugenol, IX = 15-hydroxypentadecanoic acid, X = Carpaine, XI = T-cadinol, XII = Alpha-ocimene and XIII = Hexadecanoic acid and XIV = Trimethoprim.
Discussion
Our study was designed to evaluate the bioactive compounds in Tetrapleura tetraptera (Uyayak) and in-silico assessment of the resulting bioactive compounds. The GC-MS result in our study revealed a total of 28 bioactive compounds that belong to various classes of biochemical compounds. The most abundant compound was monoterpene. In an earlier study, oxygenated monoterpenes were shown to exhibited in vitro antimicrobial and antioxidant effects against food pathogens that included E. coli. 30 This findings is in line with a review on the benefits of T. tetraptera where its benefits were highlighted to include antibacterial and antioxidant properties. 31 The bioactive compounds were further screened for their compliance with the Lipinski rule of five. Out of the twenty-eight (28) bioactive compounds, only 13 met Lipinski's ROF without any violations. According to Lipinski's ROF, an orally active drug should not violate more than one of its criteria (molecular weight < 500; ≤5 H bond donors; ≥10 H bond acceptors; and log P less than 5). 32 This implies that all 13 bioactive compounds are potential oral drug candidates.
In addition to Lipinski's ROF, we also evaluated their ADMET properties. ADMET properties are important properties that must be taken into consideration during drug discovery process. 33 Absorption of a drug is a very important consideration in drug discovery, especially for drugs intended for oral consumptio. 34 The water solubility of the ligands was higher than that of trimethoprim. Similarly, the intestinal absorption of the bioactive compounds ranged from 91% to 95% compared to that of trimethoprim, which was 71.7%. The ligands further showed variance for the evaluated distribution properties. The result of the BBB permeation indicates that all the ligands could cross the BBB compared to trimethoprim, which is predicted to poorly cross the BBB. 35 The efficacy of a lead compound depends a lot on its distribution in various tissues, which explains the lack of relationship between plasma levels and their end biological effects. 36 The metabolism of the ligands showed some consistency, as shown by the various cytochrome systems in the liver. None of the ligands, including trimethoprim, were substrates for CYP2D6, while two out of the 13 ligands were substrates for CYP3A4, implying that the majority will not be metabolized easily. However, the bulk of ligands were not inhibitors of the various cytochrome inhibitors, including CYP3A4, which are known to clear certain drugs from the system 37 further affecting their bioavailabilities. Toxicity profiling of the ligands indicates an absence of cancer risk as all the ligands were negative for Ames's test except for ligand 8 that was positive, 38 Furthermore, an absence of hepatotoxicity for all the lead compounds except the control drug was recorded. The absence of toxicity except for eugenol further indicates that these bioactive compounds can be utilized in making drugs that are safer to humans and also friendly to the environment. This is in line with Díaz et al 39 who profiled dihydrocarvone-hybrid derivatives and found them to be nontoxic. In addition to the ADMET properties, the ligands were docked against the dihydropteroate synthase of E. coli.
Molecular docking has emerged as the most utilized in silico or computational aided approach for studying interactions between proteins and other macromolecules with small lead drug compounds.15,16,38–41We docked a total of 13 ligands and trimethoprim against the dihydropteroate synthase of E. coli, and the docking scores for the various ligands ranged from −4.0 to −5.3 kcal/mol while trimethoprim gave a docking score of −6.5 kcal/mol. The higher docking score shown by trimethoprim indicates better binding with dihydropteroate synthase, even though it showed poor absorption and was predicted to be toxic. The scores shown by the ligands in our study are similar to those obtained in an earlier study with bioactive compounds from honey that were docked against the dihydropteroate synthase of S. aureus. 16 The low binding affinities observed by the ligands compared to the standard drug is in contrast to the report of Islam et al 42 who reported that limonene and its derivatives to have higher binding energies than their utilized control standard drug that ranged from −7.1 to −7.4 kcal/mol against Herpes virus indicating excellent antiviral property. However, it was further observed that the primary limonene had poor binding affinity which improved upon on the addition of functional groups 42 This implies that the low binding activities observed by the ligands in our study can be improved upon by the addition of suitable functional groups as reported by Islam et al. 43 Noumi et al, 44 in their study further showed excellent binding scores against enzymes involved in biofilm formation that were within range of our reported bioactive compounds. In addition to the docking and predicted pharmacokinetic properties of the ligands, there are reports of biological activities by these bioactive compounds.
Methone have been reported to possess antimicrobial activity against methicillin resistant Staphylococcus aureus (MRSA) via significant membrane disruption, 45 and antibacterial properties. 46 Limonene have been reported to possess excellent antibacterial activity against Listeria monocytogenes mediated via the disruption of cell wall integrity, membrane permeability and ATP synthesis. 47 Similarly, octanol derivatives such as 3,7-dimethyl-1-octanol have been shown to have possess antifungal, anthelmintic, and antioxidant activities but not cytotoxic properties. 48 Menthol have been shown to possess broad spectrum activity against Gram positive and negative bacteria (E. coli and S. aureus) via the alteration of the membrane integrity. 49 This implies that following the disruption of the membrane, a potential drug can penetrate the cell further and in the process hinder important cellular activities such as the activity of key enzymes such as dihydropteroate synthase targeted in this study and its important function of synthesizing folic acid. 49 Furthermore, Díaz et al 39 in their study established the antifungal properties of dihydrocarvone-hybrid derivatives while Noumi et al 44 in their study identified limonene and cis-dihydrocarvone with antibiofilm activity. Barrientos Ramírez et al 50 showed various derivatives of piperitone possess antibacterial activity against S. aureus and E. coli. Cordeiro et al 51 in their study revealed Terpinen-4-ol to possess remarkable antibiofilm activity even at sub-inhibitory concentrations. Eugenol have been shown to inhibit the growth of both Gram positive and negative bacteria and their biofilms. 52 On the other hand, Carpaine have been shown to have anti-plasmodial and anti-dengue activities 53 while cadinol have been shown to possess antimicrobial, antioxidant and insecticidal properties. 54 In their study, Hshemi et al showed ocimene to in-vitro antibacterial activity. 55 These findings further validate the favorable in-silico results of the bioactive compounds. Their favorable binding to dihydropteroate synthase is an indication that these bioactive compounds have the capacity to act as an antimetabolite.
The choice of dihydropteroate synthase was predicated on the fact that only prokaryotes such as bacteria elaborate this important enzyme that is involved in folic acid synthesis, while eukaryotes do not and have to rely on an external supply of folic acid since they lack this enzyme, making it an important drug target. 56 For a drug to be effective, it has to be able to get to its target. In this case, the target dihydropteroate synthase is an enzyme that belongs to the ligase class of enzymes. From the predicted targets, all the ligands had enzymes as one of their predicted targets. The bioactive compounds, despite their lower docking scores compared to trimethoprim, showed better pharmacokinetics, especially in terms of absorption and toxicity profiles.
Conclusion
Our findings indicate that T. tetraptera is rich with bioactive compounds with diverse monoterpenes and its derivatives that formed the majority of detected compounds. The next most abundant bioactive compound was sesquiterpenes while the most concentrated was lupeol which is triterpenoid ketone. Out of the 28 compounds, 13 passed passed Lipinski's ROF without any violation. Furthermore, ADMET analysis reveals favorable pharmacokinetic properties, especially the absorption, metabolism, and toxicity profiles of the bioactive compounds. The predicted target analysis, showed that the bioactive compounds could target the enzyme dihydropteroate synthase. Docking scores returned favorable binding scores and interactions that were comparable to those of trimethoprim. Put together, the findings indicate that the bioactive compounds from this edible pod, T. tetraptera, could be exploited further as potential lead compounds capable of interfering with the function of dihydropteroate synthase in E. coli.
Footnotes
Acknowledgements
We appreciate the laboratory technologists at Mifor Consult for carrying out the GC-MS.
Author's Contribution
The study was conceptualized and designed by GPB and UOE. All the microbiological analyses were handled by GPB, UOE, JCU, AAOE, and AYO (isolation, identification, and sensitivity testing). UOE, GPB, and ENM handled SMILES string generation, ADMET prediction, ligand and protein preparations, and molecular docking jointly. All the results were jointly validated by all the authors. The initial manuscript draft was done by UOE, GPB, and AYO, while the editing of the manuscript draft was carried out by all the authors. All authors took turns reading the manuscript and approved it for publication.
Availability of Data
All the data generated during the course of this study and are not captured herein will be made available on request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate
Not applicable.
Statement of Animal Rights
Not applicable as no animal subjects were utilized in this study.
Statement of Informed Consent
Not applicable as there were no human subjects.
Consent to Publication
Not applicable.