
Abstract
Introduction
Breast cancer (BRCA) is a significant public-health issue due to its prevalence, ranking as the second leading cause of cancer-related death among women both in China and worldwide. 1 Current multidisciplinary therapeutic options for BRCA, including surgery, chemotherapy, adjuvant therapy, and radiotherapy, are limited due to various effects, such as recurrence, resistance, and toxic side effects. 2 Natural mushroom-derived medicines, particularly those derived from Sanghuangporus vaninii, a large precious medicinal mushroom known as “Yanghuang” in China, are gaining more attention in BRCA treatment due to their advantages of efficiency, safety, and minimal side effects.3,4 This mushroom, which primarily grows on the trunk of Populus spp. in northeastern China, Korea, and other East Asian countries, has long been employed in the treatment of tumor-related diseases, including breast, oral, gastrointestinal, lung, and colon cancers.5,6 In recent years, there have been promising pharmacological studies on its antitumor activity, antioxidant effect, anti-inflammatory effect, uric acid lowering, etc. 7 Yu et al reported that the aqueous extracts of SV inhibited the proliferation of A375 human melanoma cells in a dose-dependent manner, and induced A375 cell cycle arrest in S phase and apoptosis by up-regulating p21 expression. 8 Wan et al studied that the polysaccharides of SV inhibited the proliferation of non-small cell lung cancer cell lines A549, 95-D and NCI-H460, especially the acidic polysaccharide affected cell morphology and colony formation in NCI-H460 cells, and its antitumor regulation via activation of the p53 signaling pathway in breast cancer MCF-7 cells.9,10 Qiu et al reported that SV extract could induce apoptosis in HT-29 cells via the intrinsic apoptotic pathway, and further found that a pyrone inoscavin A isolated from a SV extract exhibited anti-colon cancer activity via the hedgehog signaling pathway. 11 He et al reported that SV significantly inhibited the proliferation of SiHa cells. Cell cycle blockade and induction of cell apoptosis contributed to the cell death induced by SV. 12 Our recent studies have demonstrated that polyphenolic compounds from Sanghuangporus and Inonotus sp. exhibit significant antitumor activities against breast cancer and sedative effects. 13 However, the specific active components and the underlying mechanisms of their anti-proliferative effects against breast cancer remain unclear.
The comprehensive investigation of active chemical constituents and mechanisms of action is essential due to the synergistic multi-component effects exhibited by natural mushroom medicines. To achieve this objective, it is crucial to select a reasonable and efficient analytical method that can provide rapid analysis while ensuring rich and accurate data. Ultra-performance liquid chromatography quadruple detection systems are an appropriate analytical tool with high resolution, excellent sensitivity, and strong structural characterization capability. Furthermore, they enable effective separation and analysis of complex components. 14 With the rapid advancements in bioinformatics, network pharmacology has emerged as a powerful tool for exploring the multiple molecular aspects of medicine. By combining ultraperformance liquid chromatography tandem quadrupole time-of-flight mass spectrometry (UPLC-Q-TOF/MS) with network pharmacology, a systematic understanding of the active chemical constituents and their mechanisms of action can be obtained. 15
In this study, UPLC-Q-TOF/MS combined with network pharmacology was employed for rapid qualitative analysis of the chemical compositions and elucidation of anti-breast cancer mechanisms. The results will serve as a theoretical basis for further quality evaluation and application of S. vaninii.
Results
Chemical Composition
Members of the genera Sanghuangporus and Inonotus have been reported to produce a variety of yellow polyphenol pigments, known as styrylpyrones, which have shown significant biological effects. The structure of styrylpyrones contains more than 4 phenolic hydroxyl groups, which they are acidic and have a high ionization efficiency in the negative ion mode.
16
So the negative ion mode was utilized to analyze the chemical constituents of ethanol extracts of SV, and compounds were identified based on their chromatographic peak retention time, accurate relative molecular mass, MS fragmentation, as well as relevant literature. Isomers were distinguished by their characteristic MS2 fragmentation patterns reported in the literature. Figure 1 displays the full scan ESI-MS spectra in negative ion mode. Although the base peak ion (BPI) is complex, most of the chromatographic peaks are well separated. A total of 53 chemical components including 4 organic acids, 6 catechins, 13 pyranones, 13 flavonoids and 17 fatty acids were detected and identified unambiguously or tentatively. The detailed results can be found in Table 1.

Base peak ion (BPI) of ethanol extract from S. vaninii fruit body in negative ion mode.
Identification of Constituents in the Ethanolic Extract of S. vaninii by UPLC-Q-TOF-MS in Negative Ion Mode.

tR, retention times.
For SV
SV

MS (A) and MS2 (B) spectra of phelligrin C in negative ion mode.

The possible mass spectroscopic fragmentation pathway of phelligrin C in negative ion mode.
Target Identifying and Prediction
In the present study, a total of 36 polyphenols (4 organic acids, 6 catechins, 13 pyranones, and 13 flavonoids) were obtained from the fruiting body of SV using UPLC-Q-TOF/MS. By searching the Swiss Target Prediction and TCMSP databases, after eliminating duplicates, there were 415 potential targets of 33 candidate compounds, ultimately suggesting that these compounds may have common bioactive targets and connecting networks. The other three compounds SV 12, 15 and 21 were searched without targets.
Breast Cancer Differentially Expressed Genes (BRCAdegs) Search
To explore the targets for breast cancer treatment, we screened differentially expressed genes (degs) between breast cancer and normal breast tissues using the Cancer Genome Atlas (TCGA) databases. A total of 5080 BRCAdegs were discovered with filtered by FDR < 0.05, including 2989 up-regulated genes and 2091 down-regulated genes. The volcano plot represented the relationship between the significance and the fold change of BRCAdegs (Figure 4). They were highlighted by red and blue dots (|log2 (fold change)| > 1), with red representing log2 (fold change) > 1 and blue representing log2 (fold change) < 1.

The volcano diagram of BRCAdegs from TCGA. Red dots indicate up-regulated BRCAdegs, while blue dots indicate down-regulated BRCAdegs.
The Venn diagram showed the overlap of the compounds targets and BRCA-related DEGs (Figure 5), and 144 common targets were considered as potential targets of SV extracts against breast cancer.

Venn diagram of the intersection potential targets of BRCAdegs and polyphenols from S. vaninii.
Network Construction and Analysis
To explore the relationship between the common targets, the 144 intersecting targets were imported into the STRING database and Cytoscape software for network visualization analysis, and the protein–protein interaction (PPI) network had a total of 142 nodes and 1188 edges, as shown in Figure 6. Then, using the CytoHubba plug-in unit for topological analysis according to betweenness, closeness and degree, that after the intersection of the top 10 core target genes (Figure 6A-C), 8 hub genes were screened out, namely MMP9, JUN, EGF, CCND1, IL6, ESR1, FOS and EGFR. These may be key targets in the anti-breast cancer effect of SV.

Protein–protein interaction (PPI) network of total potential genes and the frequency of core genes, (A) betweenness-top10, (B) closeness-top10, (C) degree-top10.
The compound-target network was constructed using Cytoscape version 3.9.0. The network consisted of 175 nodes (31 compounds, 144 targets) and 525 interaction edges (Figure 7), resulting in an average degree of 6. In particular, the compounds with the top 6 in the compound-target network included SV26 (naringenin, degree = 45), SV27 (hesperetin, degree = 36), SV28 (sterubin, degree = 31), SV36 (phelligrin C, degree = 25), SV31 (phelligrin A, degree = 24), SV16 (phellibaumin B, degree = 24). The genes that were associated with the most compounds were CA12 (degree = 20), CA4 (degree = 18), ABCG2 (degree = 16), ESR1 (degree = 16), MMP9 (degree = 15), MMP12 (degree = 15). These high-level compounds and targets may play an important role in the anti-breast cancer effect of SV extracts.

Active compounds-targets network relationship diagram.
To further elucidate the key pathways involved in the anti-breast cancer activity, we imported the first 33 key compounds, 144 core targets, and the initial 67 enrichment pathways for SV into Cytoscape 3.9.0 software to construct a comprehensive compound-target-pathway network (Figure 8). The resulting graph comprises of 242 nodes and 1172 edges. Analysis of degree values indicated that several pathways are crucial, including pathways in cancer, neuroactive ligand-receptor interaction, chemical carcinogenesis-receptor activation, Ras signaling pathway, microRNAs in cancer, lipid and atherosclerosis metabolism, calcium signaling pathway, MAPK signaling pathway, PI3K-Akt signaling pathway AGE-RAGE signaling pathway in diabetic complications proteoglycans in cancer coronavirus disease-COVID-19 hepatitis B transcriptional misregulation in cancer human T-cell leukemia virus 1 infection IL-17 signaling pathway breast cancer Rap1 signaling pathway human cytomegalovirus infection non-small cell lung cancer. These findings demonstrate that the fruting body of SV exerts its therapeutic effects against breast cancer through multiple components acting on multiple targets via various interconnected pathways.

Active compound-target-pathway network relationship diagram.
GO and KEGG Enrichment Analysis
GO and KEGG pathway enrichment analyses were further performed to indicate the biological activity and possible mechanisms of the potential targets. The GO enrichment analysis of the central nodes produced 222 enriched GO terms (FDR < 0.05), including 134 biological processes, 34 cellular components, and 54 molecular functions. The top 15 enriched GO terms were shown in Figure 9. In detail, the top three terms in the GO biological processes were positive regulation of transcription from RNA polymerase II promoter (GO: 0045944), negative regulation of transcription from RNA polymerase II promoter (GO: 0000122), positive regulation of transcription, DNA-templated (GO: 0045893). According to the GO cell components, the terms included plasma membrane (GO: 0005886), cytoplasm (GO: 0005737), integral component of membrane (GO:0016021). The cell membrane was an essential organelle that regulated the cell signaling and controlled the nutrient transport of the cell. In terms of GO molecular functions, they were mainly enriched in identical protein binding (GO:0042802), zinc ion binding (GO:0008270), calcium ion binding (GO: 0005509).

Go analysis of potential targets of active compounds. Top 15 enriched GO terms for biological processes, molecular functions, and cellular components.
KEGG pathway enrichment analysis was conducted to further explore the possible functions of the targets (FDR < 0.05), and 67 pathways were obtained. The top 10 signaling pathways with the most number of enrichment targets were pathways in cancer (hsa05200), neuroactive ligand-receptor interaction (hsa04080), chemical carcinogenesis-receptor activation (hsa05207), Ras signaling pathway (hsa04014), microRNAs in cancer (hsa05206), lipid and atherosclerosis (hsa05417), calcium signaling pathway (hsa04020), MAPK signaling pathway (hsa04010), PI3K-Akt signaling pathway (hsa04151), as well as AGE-RAGE signaling pathway in diabetic complications (hsa04933) (Figure 10). These results indicated that the active ingredients of SV regulate multiple signaling pathways, and that signaling pathways such as MAPK and PI3K-Akt may be responsible for SV-induced growth inhibition in breast cancer cells.
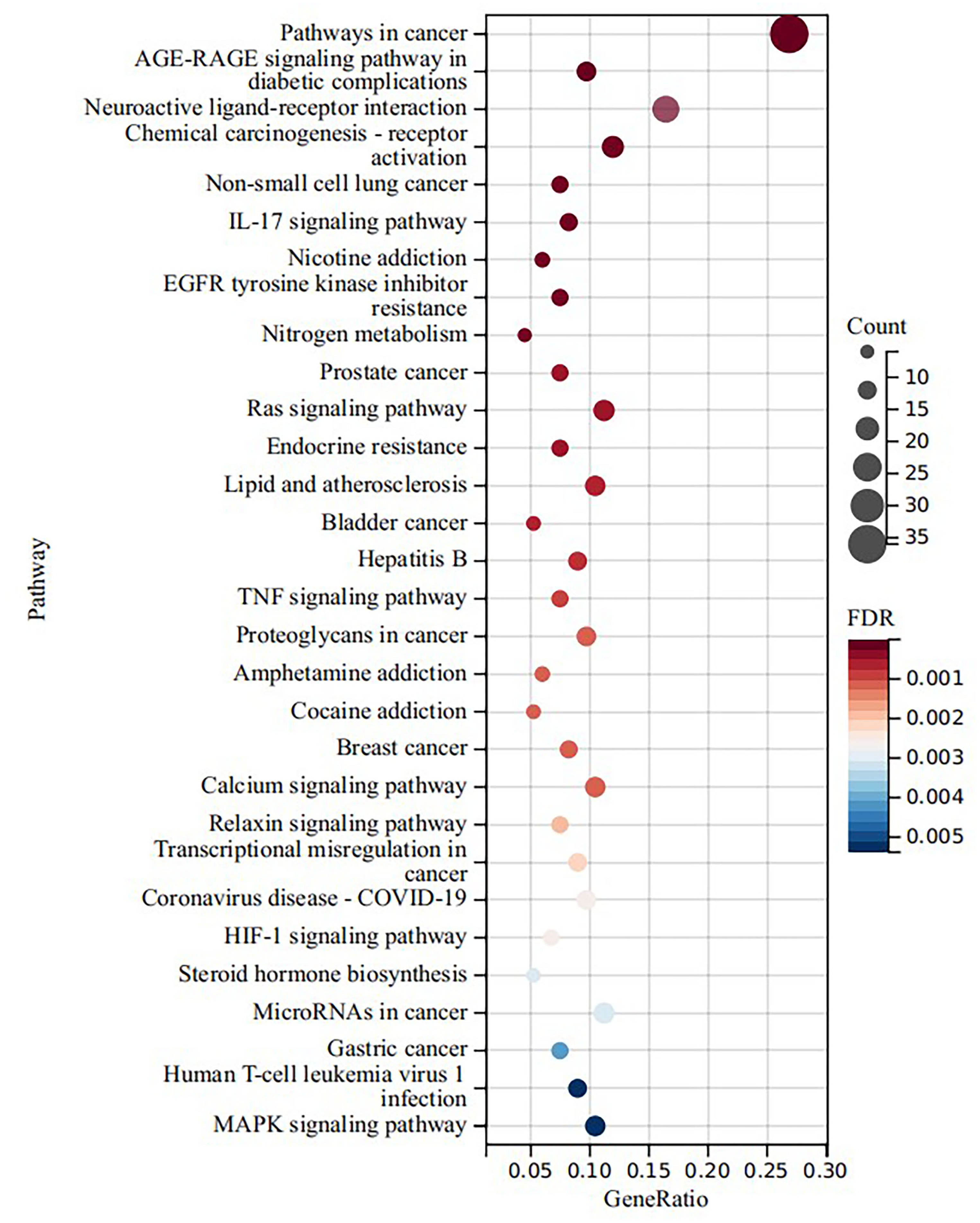
KEGG pathway enrichment analysis of potential targets of active compounds. Top 30 enriched KEGG terms.
Molecular Docking of Compounds to Hub Targets
To further validate the binding affinity between the active compounds and the key targets, molecular docking was conducted using AutoDock Tools 1.5.7 with six key compounds and eight core targets. The calculated binding free energies are presented in Table 2. The three-dimensional topological structures of the compound-target binding models were constructed, as illustrated in Figure 11. Notably, a negative binding energy indicates spontaneous binding of ligand molecules to receptor proteins. When the docking binding energy fell below the empirical threshold (−5.0 kcal/mol), it indicated stronger binding ability between the compound and target. 28 Results revealed that phellibaumin B (SV16), naringenin (SV26), hesperetin (SV27), sterubin (SV28), phelligrin A (SV31) and phelligrin C (SV36) could bind to multiple sites on MMP9 (PDB ID: 1GKC), JUN(PDB ID: 5T01), EGF(PDB ID: 1JL9), CCND1(PDB ID: 2W96), IL6(PDB ID: 1ALU), ESR1(PDB ID:4XI3), FOS(PDB ID:1FOS) and EGFR(PDBID : 1XKK) proteins with negative free energy values, indicating relatively higher affinity towards MMP9, EGF, and EGFR compared to other targets based on their respective binding energies. Moreover, one compound exhibited multi-target interactions suggesting its potential for promoting anti-breast cancer effects through multiple pathways.

Molecular docking of active components and key target proteins in breast cancer. (A) MMP9-SV31, (B) EGF-SV16, (C) EGF-SV26, (D) EGF-SV27, (E) EGF-SV28, (F) EGF-SV36, (G) CCND1-SV28, (H) EGFR-SV16, (I) EGFR-SV26, (J) EGFR-SV27, (K) EGFR-SV31, (L) EGFR-SV36.
Molecular Docking Results of the Core Antitumor Components of S. vaninii With Key Targets.

Discussion
BRCA is a complex health issue with intricate pathogenesis, a distinct lack of effective therapeutic options, and increasing morbidity. S. vaninii, a traditional medicinal mushroom used for treating tumor-related diseases, contains numerous active compounds with diverse physicochemical properties. In this work, we employed UPLC-Q-TOF/MS and network pharmacology to investigate the active compounds and mechanisms of SV in BRCA treatment. SV was considered to contain multiple compounds with different target proteins. Among these key components, phellibaumin B is a derivative of hispidin with a pyranobenzopyran moiety which has been reported by Wu et al to exhibit NF-κB inhibitory activity with IC50 value of 41.40 μM. NF-κB belongs to the nuclear factor-kappa B family and is known as critical regulators of mammalian immune and inflammatory responses for almost two decades. 21 Naringenin, hesperetin, sterubin, phelligrins C, phelligrin A are flavanone group polyphenols that possess significant pharmacological properties including anti-inflammatory, antioxidant neuroprotective hepatoprotective activities as well as anticancer effects. Naringenin exerts its anticancer effects through multiple mechanisms, including induction of apoptosis, cell cycle arrest, inhibition of angiogenesis, and modification of several signaling pathways, including the Wnt/β-catenin, PI3 K/Akt, NF-ĸB, and TGF-β pathways. 29 Hesperetin can inhibit cell proliferation, migration and BRCA stem cells, as well as induce apoptosis and cell cycle arrest in vitro. It can also inhibit tumor growth, metastasis and neoplastic changes in tissue architecture in vivo. These chemotherapeutic and chemosensitizing activities of hesperetin have been attributed to multiple mechanisms, including modulation of signaling pathways, glucose uptake, enzymes, miRNA expression, oxidative status, cell cycle regulatory proteins, tumor suppressor p53, plasma and liver lipid profiles, and DNA repair mechanisms. 30 Wu et al reported that the benzyl dihydroflavone phelligrin A plays an important role in blocking both LPS-induced and constitutive NF-κB activity in human prostate cancer cells. NF-κB-regulated genes are involved in tumor cell proliferation and metastasis, and inhibition of NF-κB signaling may be an attractive antitumor approach. 21 Phelligrin C is a new active ingredient with a structure similar to that of phelligrin A, except for the addition of 1 molecule o-hydroxytoluene. Molecular docking results showed that the binding ability of phelligrin C to the key targets EGF/EGFR was stronger. Epidermal growth factor (EGF) is a well-known growth factor that induces cancer cell migration and invasion. The EGF receptor (EGFR) and its stimulation by EGF have been implicated in a number of cell proliferative disorders or malignancies. EGFR dimerization and tyrosine autophosphorylation can activate several intracellular signaling pathways, such as PI3 K/AKT signaling pathway, JUK signaling pathway. These pathways play an important role in cell growth and proliferation. 31
PPI network analysis and KEGG enrichment revealed that the therapeutic mechanism of SV is closely related to the targets MMP9, JUN, EGF, CCND1, IL6, ESR1, FOS, and EGFR. Matrix metalloproteinase 9 (MMP9) is an enzyme that plays an important role in the first step of metastasis by degrading the extracellular matrix. 32 c-JUN is a prominent component of many AP-1 complexes and plays a critical role in the phenotype, tumorigenesis and metastasis of BRCA cells. 33 Down-regulation of IL6 expression inhibits blood vessel formation and has an immunomodulatory effect against breast cancer. 34 Acquisition of ligand-independent ESR1 mutations during aromatase inhibitor therapy in metastatic estrogen receptor (ER)-positive breast cancer is a common mechanism of resistance to hormone therapy. Preclinical and clinical studies have shown that ESR1 mutations can be pre-existing in primary tumors and enriched during metastasis. ESR1 mutations express a unique transcriptional profile that favors tumor progression, suggesting that selected ESR1 mutations may influence metastasis. 35
This study provides a preliminary revelation of the active ingredients and potential pharmacological mechanisms of SV in the treatment of breast cancer. These findings establish a vital theoretical basis and serve as a scientific reference for the future investigating the pharmacological mechanisms of SV. However, this approach is an in vitro predictive technology that lacks the ability to provide dose–response relationships, which affects the accuracy of network analysis. Therefore, their efficacy against breast cancer was further investigated in vivo and in vitro.
Conclusions
In this paper, UPLC-Q-TOF/MS technology was utilized for rapid qualitative analysis of the chemical composition of S. vaninii. As a result, 53 compounds were effectively separated and identified, including one novel compound. Thirty-three active ingredients and 144 common targets were then employed to investigate the potential mechanism by which SV exerts its effects against BRCA. The results indicated that six compounds including phellibaumin B, naringenin, hesperetin, sterubin, phelligrin A, and phelligrin C might be the core active ingredients of SV able to exert therapeutic effects against breast cancer. and the molecular mechanism may be related to targeting Ras, MAPK, PI3K-Akt, and other signal pathways associated with the key targets MMP9, EGF, and EGFR. These findings highlight the multifaceted nature of traditional Chinese fungal medicines characterized by their multi-component, multi-target, and multi-pathway properties.
Materials and Methods
Chemical Composition Analysis
Reagents and Materials
The MS-grade methanol and water were procured from Thermo Fisher Scientific Co., Ltd (Handan, China). The MS-grade formic acid was obtained from Beijing Chemical Plant (Beijing, China). For sample extraction, ethanol and water of analytical purity were used.
The wild fruiting body of S. vaninii was collected from Changbai Mountain, Jilin Province, China, and authenticated by Prof. Tolgor. A voucher specimen (No. 20220910) has been deposited at the Institute of Edible Fungi, Handan University, Handan, China.
Preparation of Sample Solution
The air-dried powdered fruiting body of SV (50 g) was subjected to ultrasonic extraction for 90 min at 45°C using 500 mL of an aqueous ethanol solution with a concentration of 80% (v/v). After centrifugation at 6000 rpm for 15 min, the supernatant was evaporated under reduced pressure at a temperature of 40 °C, and then transferred to a clean lyophilization bottle for drying purposes. The dried extracts were reconstituted with methanol and filtered through a filter with a pore size of 0.22 µm, then 10 µL of the filtrate was injected into the UPLC system for analysis.
UPLC-Q-TOF/MS Analysis
The chromatographic separation was conducted using a Waters Acquity UPLC system, which was equipped with a binary solvent system, an autosampler, and a photodiode array (PDA) detector. A temperature of 40 °C was maintained during the analysis using an Acquity UPLC BEH C18 column (2.1 mm x 150 mm, 1.7 μm, Waters, Manchester, UK). The mobile phase consisted of methanol (A) and 0.1% formic acid (Fa) in water (B), and a gradient elution procedure was employed. The following mobile phase gradient protocol was selected: 0–2 min: 5% A; 2–50 min: 5–100% A; 50–55 min: 100% A; 50–55.1 min: 100–5% A; and finally from minute 55.1 to minute 60: 5%A. Flow rate used for the analysis was set at 0.3 mL/min with an injection volume of 10 μL.
To couple the Acquity UPLC system with mass spectrometry detection, a hybrid quadrupole orthogonal time-of-flight (G2Qtof) mass spectrometer equipped with electrospray ionization (ESI) was utilized. The operating parameters were as follows: capillary voltage = 2.8 kV, sampling cone = 30 eV, extraction cone = 1.0 eV, source temperature = 100°C, desolvation temperature = 300°C, cone gas flow (N2) = 50 L/h, and desolvation gas flow (N2) = 900 L/h. The mass scan range covered m/z values from 50 to1200 Da.
Mechanism Study by Network Pharmacology
Prediction of Active Compound Targets
All polyphenols identified through screening by UPLC mass spectra were converted into canonical SMILES formats, and their potential targets were obtained from the TCMSP (https://tcmspw.com/tcmsp.php) and Swiss Target Prediction (http:// www.swisstargetprediction.ch/) databases based on the condition that probability >0.
Searching for BRCA Targets
BRCA-associated target genes were collected from the TCGA database, and RNA-Seq data from the 1185 breast cancer (BRCA) tissue samples were analyzed, including 1086 primary tumor samples and 99 normal solid tissue samples. In terms of analysis used DESeq2 data package, BRCA-related significantly differential expression genes exhibited the following characteristics: FDR < 0.05 and |log2(fold change)| > 1. Duplicate data and false-positive genes were then deleted.
Networks Construction
The SV and BRCA protein–protein interaction (PPI) networks were merged using the STRING database version 11.0 (https://string-db.org). The unconnected nodes in the network were hidden. A compound-target network and a compound-target-pathway network were constructed using Cytoscape 3.9.0 software. The compounds, targets, and pathways were described by nodes and the interaction was encoded by edges.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
GO and KEGG enrichment analyses were used to further clarify the biological interpretations of the hub genes of the network. The GO analysis included three aspects: biological process (BP), molecular function (MF) and cellular component (CC). GO terms and pathways with FDR < 0.05 were selected. In addition, a KEGG pathway network was constructed using Cytoscape version 3.9.0 software.
Molecular Docking
The 3D structures of the active compounds associated with the targets were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). The 3D structures of the target proteins were obtained from the Protein Data Bank (PDB) database (http://www.rcsb.org/). The ligand-receptor complex structures were selected and modified by removing the original ligands. The 3D structures of the ligand-receptor complexes were visualized using PyMol software. Finally, for validation of molecular docking, the preprocessed proteins and compositions were imported into Autodock Tools 1.5.7. Upon completion of all docking simulations, interactions between all active molecules and disease targets were ranked based on their strength.
Statistical Analysis
Mass spectrometry data were analyzed using Mass Lynx V4.1 software, and the structure of the compounds was predicted using the Chemspider, PubChem, and Lipidomics Gateway databases.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval is not applicable for this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by Hebei Natural Science Foundation, China (grant number C2019109043), the project of Handan University (grant number ZZ2023002) and the Funded by Science and Technology Project of Hebei Education Department (grant number ZC2023191).
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Trial Registration
Not applicable, because this article does not contain any clinical trials.
Supplemental Material
Supplemental material for this article is available online.