
Abstract
Objective
Magnolia coriacea is a rare species of Magnoliaceae and categorized as critically endangered, yet known for its valuable pharmaceutical properties. Phytochemical study of the plant was carried out, and the isolated compounds were subjected to different computational platforms to predict their chemical, inhibitory, physicochemical, and pharmacological properties.
Methods
M coriacea dried twig or leaf powder was partially extracted with n-hexane, ethyl acetate, and 90% methanol to produce the corresponding extracts, which were subjected to column chromatography. The isolated compounds were identified by nuclear magnetic resonance (NMR) spectroscopic and electrospray ionization mass spectrometry (ESI-MS) methods.
Results
Eight known aporphine alkaloids were isolated, (+)-glaucine N-oxide (
Conclusions
Isolation and molecular docking simulation of 8 aporphine alkaloids were accomplished. QSAR and ADMET regressions suggested that three compounds (
Introduction
Over the past decades, diabetes has become a health concern worldwide, particularly in middle-income countries. Notably, type 2 diabetes is characterized by insulin resistance, which accounts for 90% of all diabetic cases,1,2 resulting in reduced glucose uptake and postprandial hyperglycemia. 1 Therefore, controlling the blood-based glucose level of diabetic patients is a key strategy for reducing more serious complications such as stroke and other cardiovascular diseases.3,4
Two main strategies are commonly used to control glucose levels in blood, primarily based on the inhibition of glucose-related enzymes in the body. First, the exoenzyme α-glucosidase, extensively expressed in microbes, plants, and animal tissues, is responsible for the hydrolysis of -(1/4) and -(1/6) bonds in starch and disaccharide molecules to yield glucose. 5 A glucose-based inhibition directly reduces the blood-glucose concentration released from digestive carbohydrates.6,7 Given this significance, myriad research attempts have been invested to find potential medicines for the inhibition of enzyme activity. In particular, acarbose was the first approved substance (AGI) for an α-glucosidase inhibitory effect resulting in delayed absorption of glucose from carbohydrate-containing foods. However, this group of drugs is often known to relate to certain abdominal side efflects, eg diarrhea or flatulence.8,9 Therefore, highly effective and low-aftereffect candidates for α-glucosidase inhibition are still of interest, for which the mechanism is based upon binding to α-glucosidase thus preventing hydrolysis of the glycosidic bonds of oligo- and polysaccharides and producing the α-glucose moiety. 10 Another approach is based on protein tyrosine phosphatase 1B (PTP1B) activity. Functionally, PTP1B negatively modulates insulin signaling by taking off the phosphate moiety from essential tyrosine residues on the activated insulin receptor, inducing a reduction of glucose uptake in insulin-responsive cells. 11 PTP1B-targeted inhibition could alter the phosphorylation that leads to the insulin-responsive cell for glucose uptake. In other words, PTP1B possesses a negative regulatory effect on the insulin signaling pathway. Given this well-established knowledge, α-glucosidase 12 and tyrosine phosphatase 1B 13 can be considered antidiabetic targets of high potential.
Magnolia (Magnoliaceae) comprised approximately 210 species
14
and is often found in regions such as East and Southeast Asia, Central America (especially Mexico), and the north of South America. Many plants from this genus have been used as medicinal folk for the treatment of headache, hypertension, fever, diarrhea, and rheumatism. Magnolia coriacea (Hung T Chang & B.L.Chen), syn. Michelia coriacea, is a rare species, categorized as critically endangered (CR), level B2ab (i,ii,iii,v),
15
and endangered (EN), level B1ab (iii,v),
16
in the IUCN Red List of Threatened Species. This plant is found in China and some northern high mountain provinces in Vietnam, eg Ha Giang (Quan Ba District), Cao Bang, Tuyen Quang, and Son La,
17
as a timber tree, about 10–20 m high.
17
Phytochemical study of M coriacea confirmed the presence of essential oil, with the main components being bicyclogermacrene (12.6%) and spathulenol (17.0%)
18
and flavonol glycosides.
19
As part of our ongoing research on this species, 8 known aporphine alkaloids were successfully isolated and structurally elucidated from the twigs and leaves, ie (+)-glaucine N-oxide (
Instead of relying solely on experimental trial-and-error efforts, in silico techniques have gained increasing interest from the scientific community, especially in medical science. For various purposes, a variety of computations can be utilized in order to reach the most reliable predictions. Density functional theory (DFT) calculation is a quantum-based technique for the analysis of molecular properties, thus providing first insights into the chemical tendencies of a chemical structure. Molecular docking simulation is a classical mechanics-based method to build up inhibitory behavior, particularly of ligand–protein structures, as in this work, thus in turn reflecting the overall inhibitory effectiveness. Quantitative structure–activity relationship (QSAR) modeling can give a collective correlation between theory-based structural properties and experiment-based biological activities of certain chemical families. Among QSAR-regressed retrieval, physicochemical properties can be subjected to evaluation of biocompatibility; on the other hand, the pharmacokinetics and pharmacology of a candidate can be preliminarily viewed from the output of absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis. Altogether, the most promising chemical structures (representative input for screened candidates) can be justified for experimental attempts to specify their desirable biological activities.
In this work, evidence for the chemical composition of M coriacea extracts was collected by experimental studies, while the inhibitory potential of individual components for their diabetes-related biological assemblies was retrieved from computational analyses.
Results and Discussion
Chemistry
From the leaf and twig extracts of M coriacea, 8 aporphine alkaloids were isolated and characterized by comparison of their nuclear magnetic resonance (NMR) spectroscopic and mass spectrometry (MS) data with those in the literature:
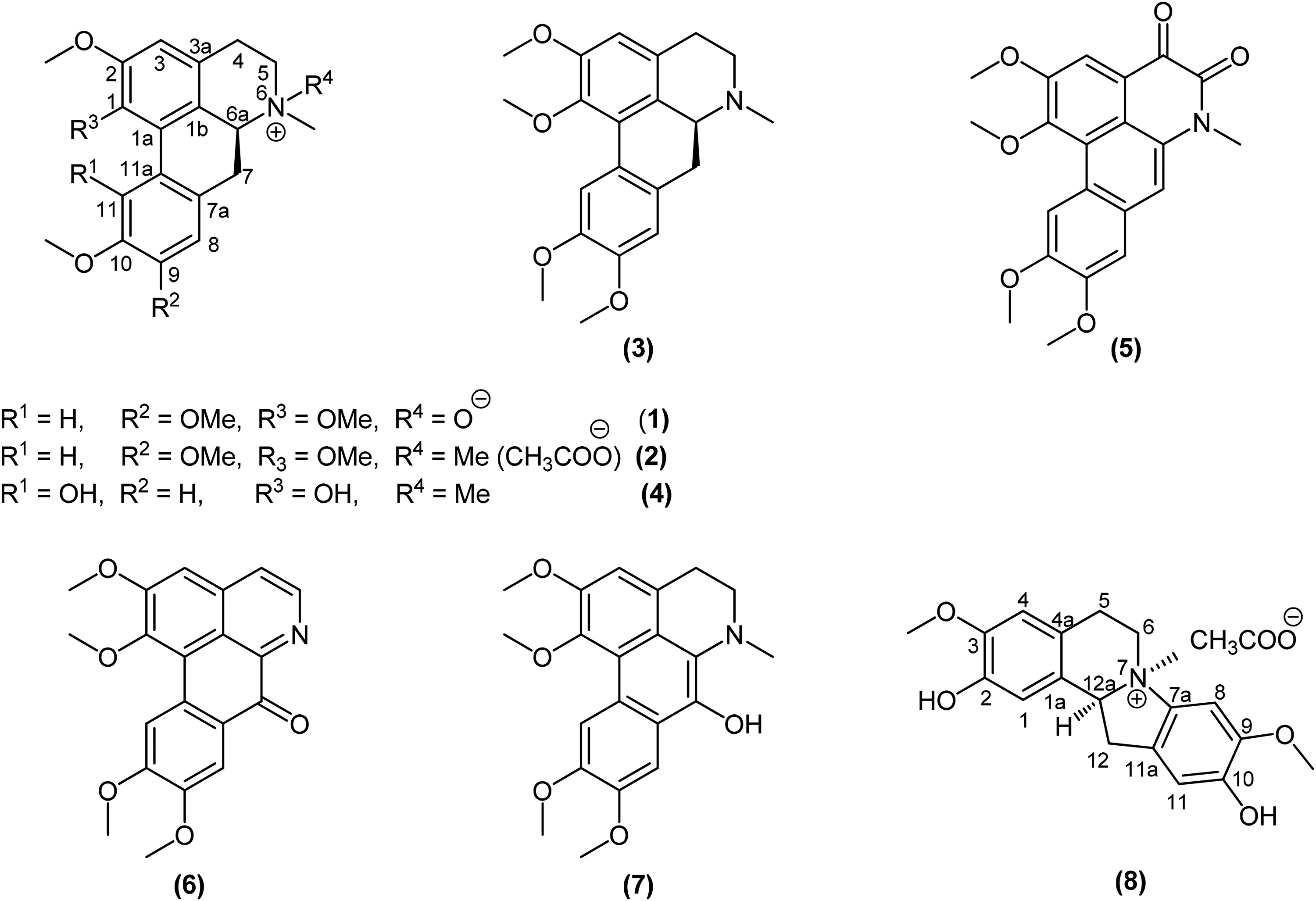
Structures of alkaloids isolated from Magnolia coriacea.
Computational
DFT-Based Chemical Properties
From the standpoint of quantum mechanics, DFT calculation can be utilized to retrieve the optimized geometry of the bioactive structures (
The data for the geometrical optimization are summarized in Table 1 and visualized in Figure 3. Overall, the self-consistent convergence reached implies stability, regarding all structures. This confirms their in-nature existence. More particularly, no abnormal constraints were detected regarding bond lengths and angles in comparison to the average characteristic values, eg 1.5 Å for C–C, 1.4 Å for C=C (aromatic), 1.4 Å for C-O, 1.2 Å for C=O, and 1.4 Å for C-N; ground state electronic energies are significantly negative, ie from −29 738.27 to −782 029.95 kcal·mol−1. All the structures considered to be polarized molecules are given their dipole moment; especially, the figures regarding

Optimized structures of
Ground State Electronic Energy and Dipole Moment Values of

The optimized geometries of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) quantum parameters are summarized in Table 2, and the corresponding electronic distributions are visualized in Figure 4. Except for

HOMO and LUMO of compounds
Quantum Chemical Parameters of Compounds

Energy gap (ΔEGAP = ELUMO−EHOMO); ionization potential I = −EHOMO; electron affinity A = −ELUMO; electronegativity χ = (I + A)/2; chemical potential µ = −χ = −(∂E/∂N)ν(r); hardness η = (I-A)/2; softness S = 1/η.
Molecular electrostatic potential (MEP) maps of the structures are given in Figure 5; by convention, reddish regions represent negativity (related to electrophilic reactivity) while bluish regions represent positivity (related to nucleophilic reactivity). Noticeably,

Molecular electrostatic potential (MEP) formed by mapping of total density over the electrostatic potential of
Overall, the results reveal the electronic behavior of the molecules, which can be used to deduce their behavior when interacting with other molecular structures.
Docking-Based Inhibition
From the standpoint of classical mechanics, molecular docking simulation can be utilized to examine the ligand–protein static behavior, thus predicting the inhibitory effectiveness of a compound toward its targeted protein structure. The docking score (DS) values and hydrophilic bonding numbers are the most important parameters; in principle, the former is the value of overall Gibbs free energy, and the latter is considered strong intermolecular bonding. Also, root-mean-square deviation (RMSD) (the average backbone-atom distances) and van der Waals interactions (hydrophobic forces) can be considered to certain extents.
In each ligand–protein duo, only the most stable intermolecular structure (in bold) is selected for more in-depth discussion since it most likely corresponds to the dominant product in reality. The susceptible sites of the biological assemblies (3W37 and PTP1B) to the ligands (

Quaternary structures of 3W37 and PTP1B with approachable sites by
Prescreening Results on Inhibitory Effect of
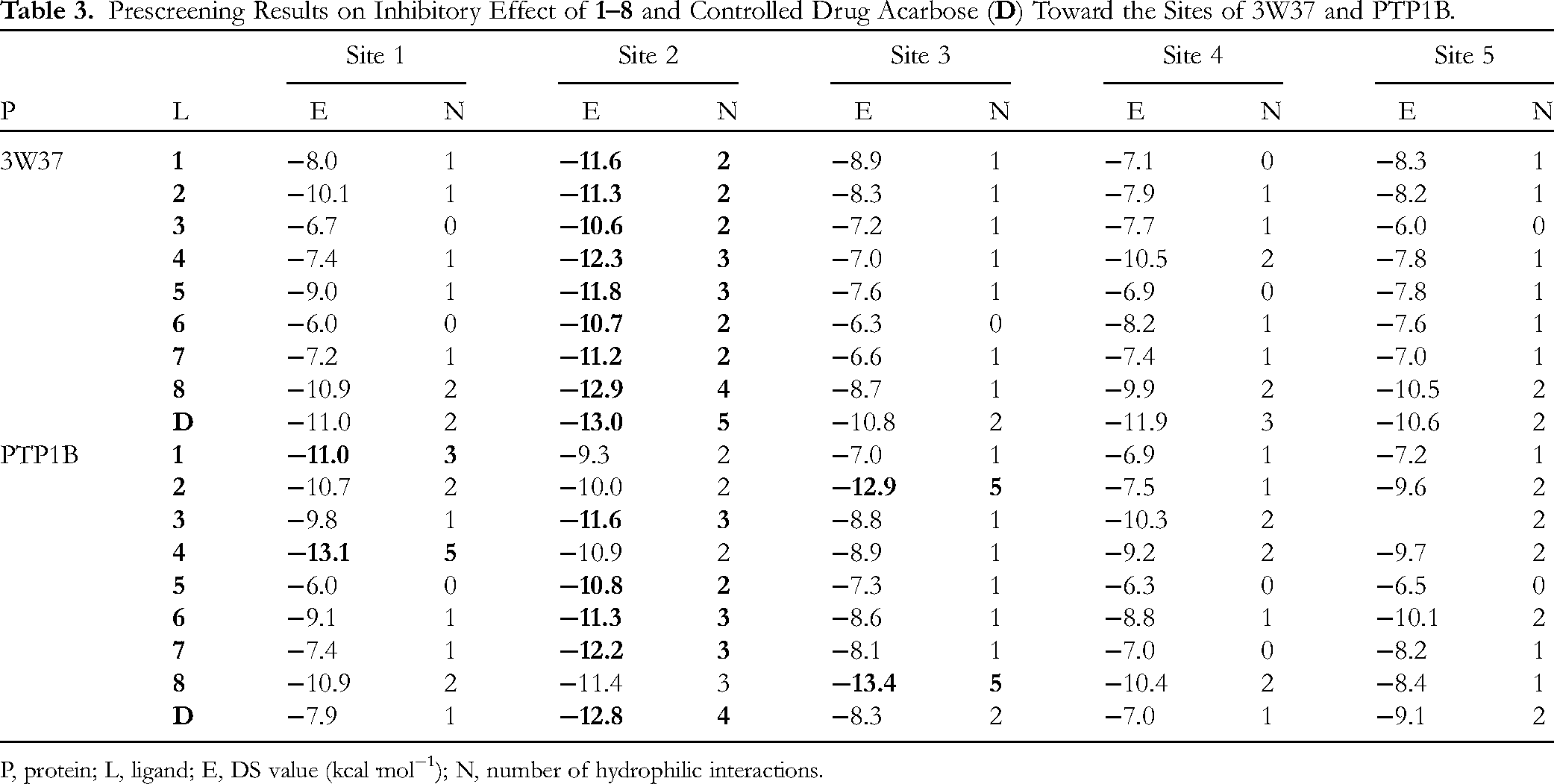
P, protein; L, ligand; E, DS value (kcal·mol−1); N, number of hydrophilic interactions.
The selected data are given in Table 4 (ligand-3W37) and Table 5 (ligand-PTP1B). According to the theoretical interpretation as given, the most effective ligand-3W37 inhibitory structures are
Molecular Docking Simulation Results for Ligand-3W37 Inhibitory Complexes.
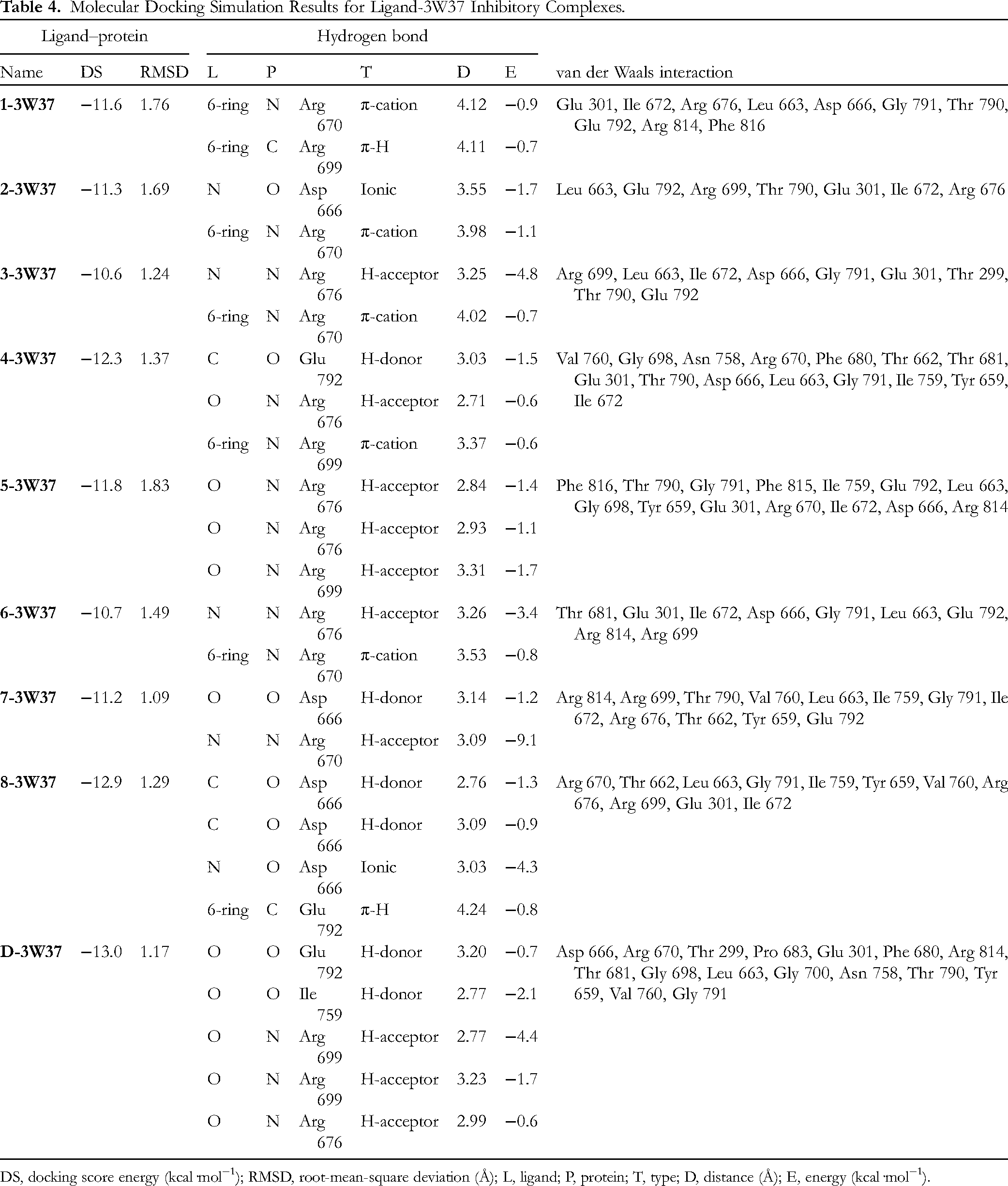
DS, docking score energy (kcal·mol−1); RMSD, root-mean-square deviation (Å); L, ligand; P, protein; T, type; D, distance (Å); E, energy (kcal·mol−1).
Molecular Docking Simulation Results for Ligand-PTP1B Inhibitory Complexes.

DS, docking score energy (kcal·mol−1); RMSD, root-mean-square deviation (Å); L, ligand; P, protein; T, type; D, distance (Å); E, energy (kcal·mol−1).
The corresponding visual projections (3D in-pose morphology and 2D interaction map) are presented in Figure 7 (ligand-3W37) and Figure 8 (ligand-PTP1B). From the scope of classical mechanics,
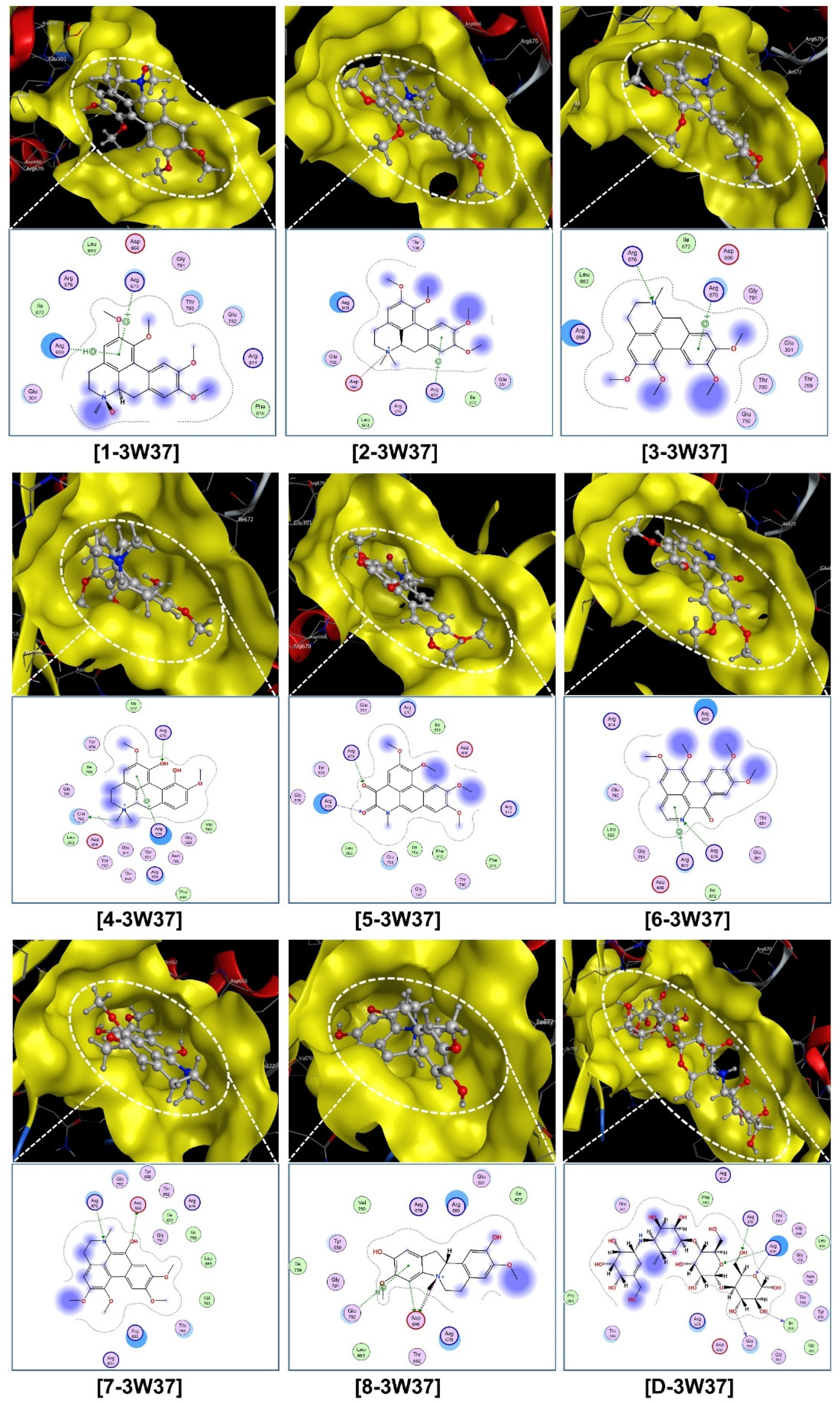
Visual presentation and in-pose interaction map of compounds
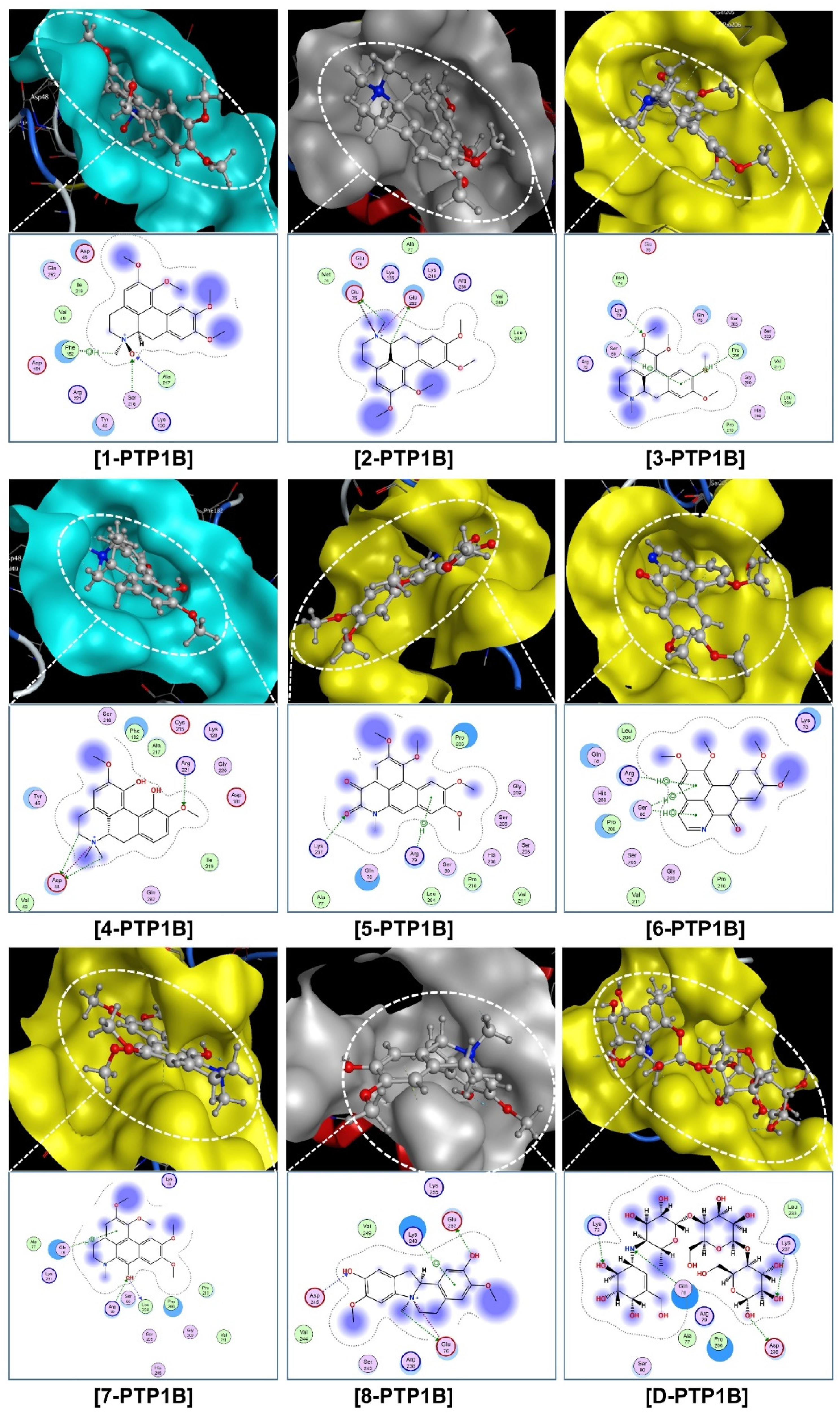
Visual presentation and in-pose interaction map of compounds
The quantum-classical consistency seen should be of great interest. The former is almost purely about the wavelike behavior of molecular electrons, while the latter is established based on the assumption that each atom in a molecule is a rigid particle and assigned with arbitrary coefficients representing its individual physical properties. However, more observations based on this approach are still needed in order to reach a solid conclusion.
QSAR-Based Physicochemical Properties
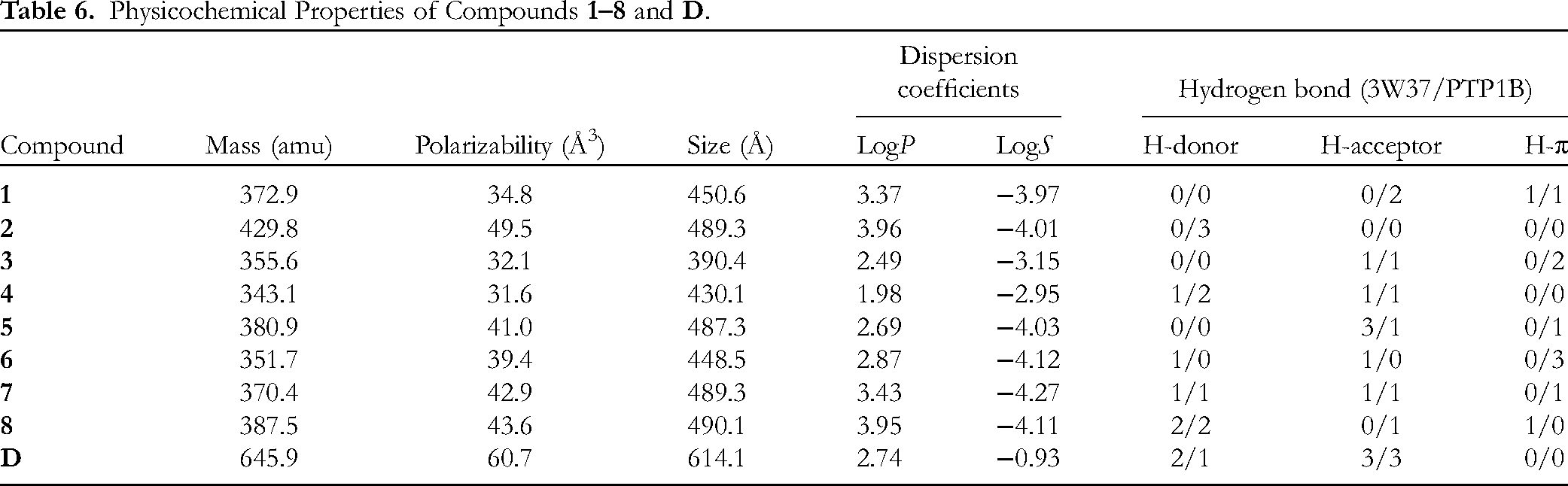
The physicochemical properties of input structures (
Physicochemical Properties of Compounds
ADMET-Based Pharmacokinetics and Pharmacology
The ADMET properties of the candidates (
ADMET-Based Pharmacokinetics and Pharmacology of Compounds

Abbreviations: BBB, blood–brain barrier; CNS, central nervous system; LOAEL, lowest observed adverse effect level; ADMET, absorption, distribution, metabolism, excretion, and toxicity.
Material and Methods
General
General experimental procedures are given in the supplementary file.
Extraction and Isolation
M coriacea dried twig powder (1.7 kg) was extracted with n-hexane, ethyl acetate, and 90% methanol (3 × 5 L each solvent, room temperature, overnight). After low-pressure concentration of the corresponding extracts, the yields obtained were n-hexane (5.5 g), ethyl acetate (28.6 g), and methanol (50 g). The methanol extract (MCR3, 50 g) was fractionated by column chromatography using gradient elution with CH2Cl2/MeOH (100/0 to 50/50, v/v), then with CH2Cl2/MeOH/H2O (50/50/0.1% to 0/90/10, 0/80/20, and 0/50/50 v/v), and finally MeOH/H2O/AcOH (50/50/0.1% to 0/100/0.1%) to provide 33 fractions (MCR3.1→MCR3.33). Fraction MCR3.12 (50.5 mg), after MeOH-based recrystallization, gave
Computation
Quantum Chemical Calculation
Molecular geometrical and electronic structures (of
Molecular Docking Simulation
Ligand–protein interactability was predicted by a docking-based technique using MOE 2015.10, typically, following four main steps.42,43,44
Step 1 Sources for simulation input: data for biological assemblies of α-glucosidase and tyrosine phosphatase 1B (PTP1B) were retrieved from Worldwide Protein Data Bank (Entry ID: PDB-3W37; DOI: 10.2210/pdb3W37/pdb) and UniProtKB (Entry ID: UniProtKB-A0A0U1XP67), respectively. Chemical formulae ( Protein-active region specification: active range (between ligand and amino acids) was defined within 4.5 Å. Experiment-based attached residues, if any, were deleted. Configuration: strength-5000 tether–receptor method; refinement 0.0001 kcal·mol−1 Å−1; format *.pdb. Ligand optimization: structural abnormalities (if any) of the ligands were checked. Configuration: energy-minima Conj Grad method; termination 0.0001 kcal·mol−1; allowed interactions 1000; empirical charge-assigning Gasteiger–Huckel method. Ligand–protein inhibitory interaction was simulated. Configuration: selected poses 10; iterative solutions 1000; fragmental solutions 200; format *.sdf. The ligand molecules were removed from the converged ligand–protein structures and then docked back. Validity conditions: (1) all RMSD values < 1.5 Å and (2) no significant differences (between corresponding systems) in RMSD value. In principle, DS value represents a pseudo-Gibbs free energy, thus serving as the primary indicator for the stability of a ligand–protein structure or, in other words, its inhibitory effectiveness.
Step 2. Docking simulation.
Step 3. Benchmarking simulation.
Step 4. Parameter interpretation.
Figure 1 presents the referenced structures (α-glucosidase and tyrosine phosphatase 1B) and the control drug (acarbose). Those for the ligands are given in Figure 2 (after the discussion for structural elucidation).

Crystal structures of (A) α-glucosidase (PDB-3W37; DOI: 10.2210/pdb3W37/pdb), (B) tyrosine phosphatase 1B (PTP1B; ID: UniProtKB-A0A0U1XP67), and (C) structural formula of controlled drug, acarbose (
QSAR-Based Analysis
Druglikeness properties of phytochemicals were predicted by reference of QSAR-derived physical properties (based on the Gasteiger–Marsili method 45 ) to Lipinski's rule of 5: 46 the former output molecular mass (Da), polarizability (Å3) and size (Å), and dispersion coefficients (logP and logS); on the other side, the rule set criteria for a good membrane-permeable candidate, ie (1) molecular mass < 500 Da, (2) hydrogen bond donors ≤ 5, (3) hydrogen bond acceptors ≤ 10, and (4) logP < +5.47,48 As a result, oral pharmacological suitability, in particular, and biological compatibility, in general, can be drawn out.
ADMET-Based Analysis
ADMET properties of the input structures (
Conclusions
This study reports for the first time the isolation of aporphine alkaloids from M coriacea and examines their diabetes-related potential based on computational models. The leaf and twig extracts contain 8 aporphine alkaloids: (
Supplemental Material
sj-docx-1-npx-10.1177_1934578X231176926 - Supplemental material for In Silico Investigation of Aporphine Alkaloids Isolated From Magnolia coriacea Against α-Glucosidase and Tyrosine Phosphatase 1B
Supplemental material, sj-docx-1-npx-10.1177_1934578X231176926 for In Silico Investigation of Aporphine Alkaloids Isolated From Magnolia coriacea Against α-Glucosidase and Tyrosine Phosphatase 1B by Pham Thi Ninh, Thanh Q. Bui, Nguyen Thi Dung, Tran Van Sung, Tran Thi Phuong Thao, Chu Thi Thu Ha, Bui Van Thanh, Tran Van Chien, Phan Tu Quy, Nguyen Thanh Triet, Nguyen Minh Thai and Nguyen Thi Ai Nhung in Natural Product Communications
Footnotes
Declaration of Conflicting Interests
The author(s)declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Vietnam Academy of Science and Technology (grant number VAST04.09/20–21). This research was funded by Hue University (grant number DHH2022-01-198). The authors also acknowledge the partial support of Hue University under the Core Research Program, Grant No. NCM.DHH.2020.04.
Informed Consent
There are no human subjects in this article, and informed consent is not applicable.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.