
Abstract
Background:
Sasanlimab (PF-06801591), a humanized immunoglobulin G4 monoclonal antibody, binds to programmed cell death protein-1 (PD-1), preventing ligand (PD-L1) interaction.
Objectives:
To evaluate pharmacokinetics (PK), safety, tolerability, and efficacy of two subcutaneous sasanlimab dosing regimens.
Design:
An open-label study consisting of phases Ib and II. Phase Ib: non-randomized, dose escalation, and expansion study in Asian participants with advanced malignancies.
Phase II:
conducted globally in participants with non-small-cell lung cancer with PD-L1 positive or PD-L1 status unknown tumors; participants were randomized 1:2 to receive subcutaneous sasanlimab 300 mg once every 4 weeks (300 mg-Q4W) or 600 mg once every 6 weeks (600 mg-Q6W).
Methods:
Primary endpoint in phase Ib: dose-limiting toxicity (DLT) occurring in first treatment cycle; in phase II: Ctrough and AUC.
Results:
A total of 155 participants (phase Ib, n = 34; phase II, n = 121) received sasanlimab. Phase Ib: no DLT reported. Phase II: ratio of adjusted geometric mean for AUCtau was 231.2 (90% CI, 190.1–281.2) and Ctrough was 111.5 (90% CI, 86.3–144.0) following 600 mg-Q6W (test) versus 300 mg-Q4W (reference). Phase Ib: grade 3 treatment-related adverse events (TRAEs) occurred in 1/4 (25%) and 3/12 (25%) participants treated in 300 mg-Q4W dose escalation and expansion cohorts, respectively. Phase II: grade 3 TRAEs occurred in 3/41 (7.3%) and 3/80 (3.8%) participants treated with 300 mg-Q4W and 600 mg-Q6W, respectively; no grade 4/5 TRAEs. Phase II: confirmed objective response was observed in 11/41 (26.8% (95% CI, 14.2–42.9)) and 12/80 (15.0% (95% CI, 8.0–24.7)) participants treated with 300 mg-Q4W and 600 mg-Q6W, respectively.
Conclusions:
Phase Ib regimens were considered safe with no DLTs reported. In phase II, 600 mg-Q6W regimen criteria were met for AUCtau and Ctrough metrics to support PK-based extrapolation of efficacy of alternative regimen. Regimens were well tolerated, showing anti-tumor activity in participants with advanced solid tumors. Administration of sasanlimab at a dose of 600 mg-Q6W subcutaneously may serve as a convenient alternative to 300 mg-Q4W administration.
Trial registration:
NCT04181788 (ClinicalTrials.gov); 2019-003818-14 (EudraCT).

Keywords
Background
Immune-mediated control of tumor growth is mainly regulated by interactions between programmed cell death protein 1 (PD-1)—an inhibitory receptor expressed on the surface of T cells 1 —and its ligand programmed cell death 1 ligand-1 (PD-L1) in the tumor microenvironment.2–4 Intravenous anti-PD-1/PD-L1 agents, such as nivolumab and pembrolizumab, have demonstrated anti-tumor survival benefits and are widely used in multimodal cancer treatment strategies for patients with various malignancies, including non-small-cell lung cancer (NSCLC), malignant melanoma, and renal cell carcinoma.3,5 The desirability of alternative treatment administration methods that improve patient experience and decrease costs has prompted the evaluation of subcutaneous (SC) anti-PD-1/PD-L1 agents.6,7
Sasanlimab (PF-06801591) is a humanized, hinge region-stabilized immunoglobulin G4 monoclonal antibody with antagonistic activities specific to human PD-1. It can selectively and reversibly bind with high affinity to human PD-1, forming a stable complex that blocks its interaction with PD-L1 and PD-L2. In vitro, sasanlimab increased T-cell proliferation and cytokine secretion (interferon-γ and interleukin-2) when PD-L1 is highly expressed. Sasanlimab accelerated the incidence of GvHD through T-cell proliferation and cytokine secretion in a xenogeneic model of acute GvHD and halted MC-38 colon adenocarcinoma tumor growth in human PD-1 knock-in mice.5,7–9 Sasanlimab blockade of the interaction between PD-1 on T cells and its ligands on tumor cells is expected to restore anti-tumor immunity and forms the basis for an immunotherapeutic approach to treat cancer. In a first-in-human trial, participants received sasanlimab by SC administration and the recommended clinical dose of sasanlimab for participants with advanced solid tumors was selected as 300 mg SC every 4 weeks (Q4W). This regimen was shown to be tolerable and have anti-tumor activity. 7 Furthermore, in a phase Ib/II dose expansion study in patients with locally advanced or metastatic NSCLC and urothelial carcinoma, patients received 300 mg of sasanlimab SC Q4W. This dose was well tolerated with promising clinical benefits. 10
According to current United States Food and Drug Administration recommendations, alternative dosing regimens for anti-PD-1/PD-L1 antibodies can be derived using a pharmacokinetic (PK)-based model-informed drug-development approach, if both the area under the plasma concentration-time curve (AUC) and the concentration of drug at the end of the dosing interval (Ctrough) at the steady state for the test regimen are no more than 20% lower compared with the parameters of the reference dosing regimen used to establish efficacy in clinical trials (i.e., 300 mg Q4W for sasanlimab). 11 However, if geometric mean Cmax of the test regimen is >25% of the reference regimen, additional evidence to assess the safety of the alternative regimen would be required. 11
Objective
The objective of this phase Ib/II study (EudraCT:2019-003818-14; NCT04181788) was to evaluate the PK, safety profile, and anti-tumor activity of an alternative dosing regimen of SC sasanlimab in participants with advanced malignancies in phase Ib and NSCLC in phase II.
Design
This was an open-label study consisting of two phases, phase Ib and phase II, conducted at 28 sites in parallel. These phases were conducted in parallel to expedite phase II evaluations outside Japan and China, facilitate phase I trials in Japan and China for future new drug application (NDA) purposes, and potentially allow Japanese participants to transition into phase II. Evaluations for countries other than Japan and China were not delayed due to the prior dose escalation carried out in a previous study (B8011001 study [NCT02573259]). Phase Ib was composed of two parts: dose escalation and dose expansion in Asian participants with advanced malignancies from Japan and China. Phase II was conducted in participants with NSCLC in Taiwan, South Korea, Ukraine, and Russia. Participants from Japanese sites were allowed to enter the global phase II part after the phase Ib dose escalation part was completed. Participants from Chinese sites only joined the phase Ib expansion part of the study due to late-opening.
In phase Ib, participants in arm A1 received sasanlimab SC 300 mg Q4W and participants in arm B1 received sasanlimab SC 600 mg Q6W (Figure 1). In phase II, participants with advanced or metastatic NSCLC were randomized 1:2 to receive sasanlimab SC 300 mg Q4W (arm A2) or sasanlimab SC 600 mg Q6W (arm B2; Figure 1). The starting dose (300 mg Q4W) was determined based on the potential clinical benefit seen in previous studies.7,10 Randomization was stratified by line of therapy (first line vs second line) for advanced or metastatic NSCLC. The study used interactive response technology (IRT) for participant randomization/treatment allocation in phase II.
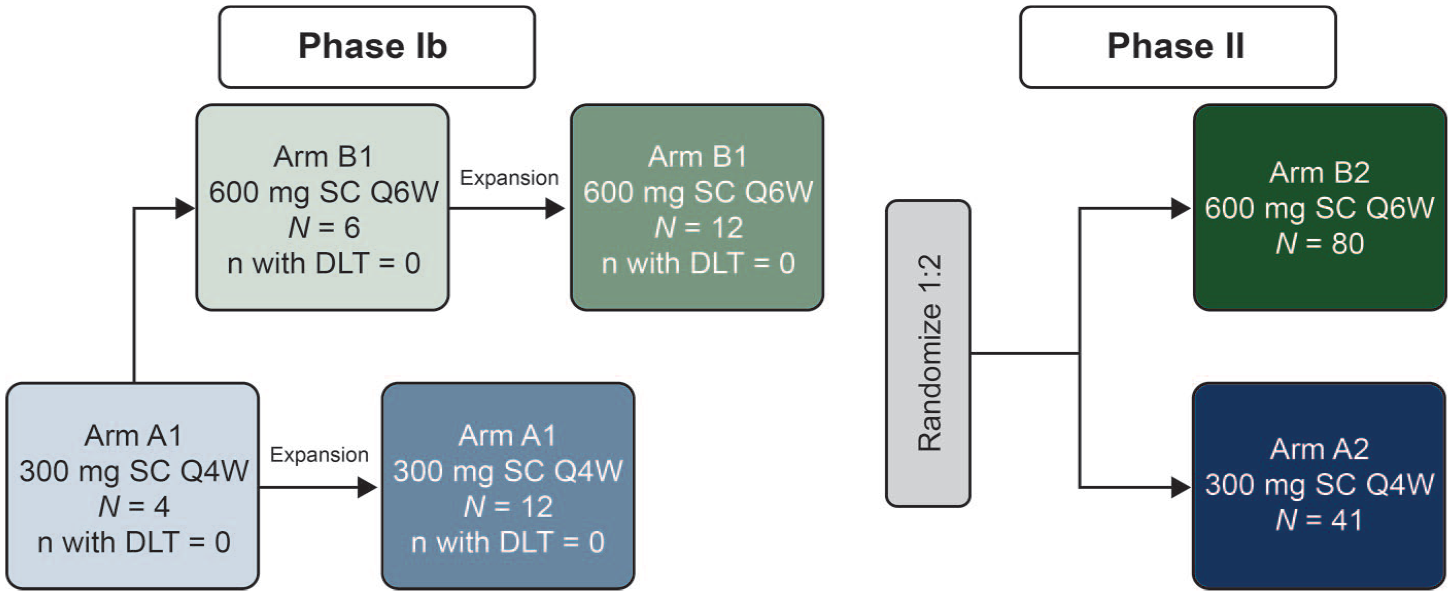
Study design.
Treatment duration was defined as the number of weeks between the first and last dose plus 4 weeks or 6 weeks for the Q4W or Q6W dosing regimen, respectively.
Methods
This study is reported in line with the CONsolidated Standards Of Reporting Trials-Dose-finding Extension (CONSORT-DEFINE) statement (Supplemental Appendix A). 12
Endpoints
In phase Ib, the primary endpoint was dose-limiting toxicity (DLT) occurring in the first cycle of treatment, defined as the time from the first dose to the next expected dose of sasanlimab (4 weeks in arm A1 and 6 weeks in arm B1). A DLT was defined as any grade 5 adverse event (AE) not due to the underlying disease or extraneous causes; prespecified grade 3/4 hematologic toxicity (i.e., grade 4 neutropenia lasting >7 days; febrile neutropenia; grade ⩾3 neutropenic infection; grade 4 thrombocytopenia or grade 3 thrombocytopenia with significant bleeding or requiring medical intervention; grade 4 anemia or grade 3 anemia requiring transfusion or steroids); prespecified grade 3/4 non-hematologic toxicity (i.e., any grade 4 non-hematologic AE; grade 3 AE lasting >3 days despite optimal supporting care; grade 3 central nervous system AE regardless of duration; AE meeting criteria for drug-induced liver injury); or a delay of ⩾3 weeks in receiving next scheduled dose due to any persisting treatment-related toxicities.
In phase II, the primary endpoints were the PK parameters Ctrough and AUC from time zero to the end of the dosing interval (AUCtau) at steady state at 12 weeks, where intensive PK was collected from each regimen across the respective interval beginning with the dose administered at 12 weeks to calculate AUC exposure. The PK parameters in phase Ib were set as secondary endpoints to assess any effect of Japan or China participation into further global studies or ethnicity on sasanlimab pharmacokinetics.
Secondary endpoints for safety in both phase Ib and phase II included AEs as graded by National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0 and laboratory test abnormalities. Secondary endpoints for efficacy for both phase Ib and phase II studies were confirmed objective response (OR) and time to response (TTR) assessed by the investigator according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Secondary endpoints for immunogenicity for both phase Ib and phase II included the incidence and titer of anti-drug antibody (ADA) and neutralizing antibody (NAb) levels. PD-L1 expression at baseline was assessed as a pharmacodynamic secondary endpoint to allow the assessment of the potential correlation between clinical activity and PD-L1 expression in baseline tumor tissue. PD-L1 status at baseline was considered high if ⩾25% of tumor cells exhibits membrane staining; low if <25% of tumor cells exhibits membrane staining; and unknown if the participant did not have the corresponding data collected; further details about the PD-L1 status assessment can be found below.
PD-L1 status assessment in tumor tissue
Baseline tumor tissue was obtained from biopsy or surgery before study enrollment to identify patients most likely to benefit from treatment. Where such tumor biospecimens were not available, a de novo biopsy from a locally recurrent or metastatic tumor site was performed at screening. Baseline PD-L1 protein expression level was assayed by using immunohistochemistry method at a central laboratory using Ventana SP263 antibody. End-of-treatment biopsies were also obtained from participants who permanently discontinued treatment due to disease progression, except where this posed an unacceptable risk to the participants.
Imaging tumor response assessment was conducted every 12 weeks (±7 days) from cycle 1 day 1 for both regimens until progressive disease assessed by the investigator (using RECIST v1.1), withdrawal, or start of new cancer therapy.
Treatment-related adverse events (TRAEs) were those events with onset dates occurring after the first dose of study treatment through the earliest of either 30 days after date of last dose of study treatment or start date of new anticancer drug. Blood samples (3 mL) for PK assessments were taken on days 1, 8, 15, and 22 for cycles 1 and 4, and on day 1 of all other cycles. Blood samples (6 mL) for ADA and NAb assessments were taken at baseline (cycle 1 day 1 predose) and on day 1 of cycles 2, 3, 4, 6, 8, 10, and at the end of treatment for Q4W dosing regimen or at baseline (cycle 1 day 1 predose) and on day 1 of cycles 2, 3, 4, 5, 7, and at the end of treatment for Q6W dosing regimen.
Participant population
Phase Ib enrolled Asian participants only, and phase II enrolled global participants. For phase Ib and phase II, participants were 18 years or older (20 years or older in Japan; 19 years or older in South Korea) with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, an estimated life expectancy of at least 3 months and adequate bone marrow, and renal and liver function (Supplemental Appendix B).
In phase Ib, participants required histological or cytological diagnosis of advanced solid tumor with clinical evidence of response to anti-PD-1 or PD-L1 agents although no PD-L1 testing was required for study inclusion and the use of prior anti-PD-1/PD-L1 agents was allowed. Additionally, participants should have received at least one prior line of therapy for recurrent or metastatic disease and must have progressed/relapsed, be refractory, or intolerant to standard therapy approved for the specific tumor type.
In phase II, additional inclusion criteria required that participants had documented diagnosis of stage III NSCLC and were not eligible for surgical resection or definitive chemoradiation or had stage IV NSCLC per International Association for the Study of Lung Cancer classification; had no prior therapy with PD-1/PD-L1 agents; had at least one measurable lesion defined by RECIST version 1.1 that was not previously irradiated. Phase II participants with tumors known to be PD-L1 positive (Tumor Proportion Score [TPS] ⩾1%) or with tumors with unknown PD-L1 status were eligible (PD-L1 testing was not required for participants with unknown status). In addition, participants may have received up to one line of prior therapy in advanced or metastatic disease settings. Furthermore, previous treatment toxicities and resolutions were considered during the randomization process through the inclusion of following criterion: resolved acute effects of any prior therapy to baseline severity or Common Terminology Criteria for Adverse Events grade ⩽1 except for AEs not constituting a safety risk by investigator judgment.
Exclusion criteria in both phases were symptomatic brain metastases requiring steroids; interstitial lung disease history or complication; or vaccination with live attenuated vaccines 4 weeks before randomization. An additional exclusion criterion for phase Ib participants was history of a grade ⩾3 immune-mediated AE that was considered related to prior immune modulatory therapy and required immunosuppressive therapy. Participants were excluded from phase II enrollment if they had an EGFR mutation, a BRAF mutation, or a known ALK or ROS1 translocation/rearrangement.
Treatment discontinuation may occur for the following reasons: objective disease progression, global deterioration of health status, unacceptable toxicity, pregnancy, significant protocol violation, lost to follow-up, participant refused further treatment, study terminated by sponsor, or death. It is important to note that discontinuation of study treatment does not represent withdrawal from the study.
Statistical analyses
The primary analysis included all data up to the clinical cutoff date of September 7, 2022; this corresponds to 8 months after the last participant’s first dose in phase Ib and 17 months after the last participant was randomized in phase II. For the primary PK analysis in phase II, it was projected that a sample size of 90 PK-evaluable participants (30 and 60 participants for 300 mg Q4W and 600 mg Q6W arms, respectively) would provide at least 80% power. This would ensure that the lower bound for the 90% confidence interval (CI) for the ratio of the 600 mg Q6W to 300 mg Q4W treatment regimen for the geometric mean of AUCtau and Ctrough at steady state would be at least 80%. This also used an assumption that the coefficient of variation is 26% for AUCtau and 40% for Ctrough, and the true ratio is 1.0 for both endpoints.
All efficacy analyses were performed based on dose levels assigned at the time of enrollment for phase Ib and phase II using the full analysis set (defined as all enrolled participants who received at least one dose of sasanlimab in phase Ib, and all participants who were randomized in phase II). Efficacy endpoints of OR and TTR were summarized based on investigator assessment using RECIST v1.1. OR was defined as a confirmed complete response (CR) or partial response (PR) according to RECIST version 1.1 based on investigator assessment, from the date of first dose of study treatment (phase Ib) or randomization (phase II) until the date of the first documentation of disease progression, start of new anticancer therapy, or death, whichever is earlier. The OR rate (ORR), defined as the proportion of participants in the analysis population with OR, was calculated along with the two-sided 95% CI using the Clopper–Pearson method. TTR was defined for participants with confirmed OR (CR or PR) as the time from the date of the first dose (phase Ib) or randomization (phase II) to the date of the first documentation of objective tumor response, which was subsequently confirmed. TTR was summarized using simple descriptive statistics (mean, standard deviation, median, min, max, Q1, Q3).
For comparison of exposure metrics, AUCtau and Ctrough at 12 weeks were selected as steady state indicators. Comparison of the PK profile at steady state and after the first dose would account for any time-dependent exposure changes, if observed.
Results
Participant demographics and baseline disease characteristics
Between April 2020 and January 2022, 155 participants (phase Ib, n = 34; phase II, n = 121) were enrolled and treated with sasanlimab in the study (Table 1). In phase Ib, 16 participants received sasanlimab 300 mg Q4W (dose escalation, n = 4; dose expansion, n = 12) and 18 participants received sasanlimab 600 mg Q6W (dose escalation, n = 6; dose expansion, n = 12). In phase II, 41 participants were randomized to and received sasanlimab 300 mg Q4W and 80 participants were randomized to and received sasanlimab 600 mg Q6W.
Participant demographics and baseline disease characteristics in phase Ib and phase II (full analysis set).

AJCC, American Joint Committee on Cancer; CRF, case report form; ECOG, Eastern Cooperative Oncology Group; IRT, interactive response technology; N, total number of participants; n/a, not applicable; NSCLC, non-small-cell lung cancer; PD-L1, programmed cell death 1 ligand 1; Q4W, every 4 weeks; Q6W, every 6 weeks; SC, subcutaneous; SD, standard deviation; TNM, tumor, nodes, metastases.
Phase Ib: advanced solid tumors with clinical evidence of response to anti-PD-1/PD-L1 agent (no PD-L1 testing was required). Phase II: advanced or metastatic NSCLC.
In phase Ib, the following tumor types were enrolled: adrenal gland cancer (n = 1), anal squamous cell carcinoma (n = 1), cervix carcinoma (n = 2), cholangiocarcinoma (n = 1), hepatocellular carcinoma (n = 1), lung neoplasm malignant (n = 2), malignant melanoma (n = 6), nasopharyngeal cancer (n = 1), NSCLC (n = 8), osteitis deformans (n = 1), ovarian cancer (n = 4), renal cell carcinoma (n = 4), small intestine carcinoma (n = 1), transitional cell carcinoma (n = 1).
Phase Ib: participant must have received at least one prior line of therapy for recurrent or metastatic disease and must have progressed/relapsed, be refractory, or intolerant to standard therapy approved for the specific tumor type. Phase II: up to one line of prior therapy in advanced or metastatic disease settings but participants should not have received prior treatment with anti-PD-1/PD-L1 drugs.
PD-L1 status is considered high if ⩾25% of tumor cells exhibit membrane staining; low if the criteria for PD-L1 high status is not met; and unknown if the participant in biomarker analysis set does not have the corresponding data collected.
The median age for all enrolled participants (phase Ib and phase II) was 63 years, and more participants were men (74.8%) than women (25.2%; Table 1). Most participants had stage IV disease (89.7%). Overall, 52.3% of study participants had received at least one prior anticancer drug therapy for advanced disease, and 16.1% and 20.0% of the participants had received at least one prior anticancer radiotherapy and/or at least one prior anticancer surgery, respectively (Table 1). Participants in phase II were stratified for randomization by line of therapy (by IRT), with 57.9% overall stratifying sasanlimab as a first-line therapy and 42.1% overall stratifying sasanlimab as the second-line therapy for advanced or metastatic disease (Table 1).
Treatment duration
In phase Ib dose escalation, the median duration of treatment was 17.0 and 12.3 weeks for sasanlimab 300 mg Q4W and 600 mg Q6W, respectively (Supplemental Table 1). In phase Ib dose expansion, median treatment duration of sasanlimab 300 mg Q4W and 600 mg Q6W was 16.0 and 36.4 weeks, respectively. In phase II, median treatment duration was 24.3and 31.4 weeks for sasanlimab 300 mg Q4W and 600 mg Q6W, respectively.
Dose reductions were not permitted. There was no apparent difference in the frequency of dose delays across phase Ib and phase II ranging between 16.7% and 25.0% in the 300 mg Q4W dosing regimen and between 16.7% and 33.3% in the 600 mg Q6W dosing regimen (Supplemental Table 1).
Pharmacokinetics of sasanlimab
The PK profile of sasanlimab was assessed after 300 mg Q4W and 600 mg Q6W single administration on cycle 1 and at steady state (cycle 4 for 300 mg Q4W and cycle 3 for 600 mg Q6W) in phase Ib and phase II. The Tmax after repeat dosing was observed at the first planned sampling time (168 hours post dose) for both regimens. After repeat dosing of 300 mg Q4W and 600 mg Q6W, the effective half-life was 31.3 and 27.3 days, respectively, for the combined phase Ib and phase II populations.
Exposure metrics observed in phase Ib in Asian participants (including dose escalation and expansion), and in phase II in global participants, showed a high degree of overlap, indicating the lack of regional or ethnicity differences (Supplemental Table 2). Box and whisker plots of the individual serum sasanlimab steady-state Cmax and Ctrough values are shown in Supplemental Figures 1 and 2.
In analysis from phase II data, the adjusted means AUCtau, for sasanlimab were: 600 mg Q6W, 48,090 µg.h/mL; 300 mg Q4W, 20,800 µg.h/mL, with a ratio (90% CI) of 231.2 (190.1–281.2). Adjusted means for AUC normalized over the dosing interval across regimens (Cavg) were: 600 mg Q6W, 47.7 µg/mL; 300 mg Q4W, 31.0 µg/mL; the ratio (90% CI) was 154.2 (126.7–187.5); Ctrough adjusted means were: 600 mg Q6W, 24.0 µg/mL; 300 mg Q4W, 21.5 µg/mL; the ratio (90% CI) was 111.5 (86.3–144.0). Cmax values were: 600 mg Q6W, 76.5 µg/mL; 300 mg Q4W, 41.7 µg/mL; the ratio (90% CI) was 183.5 (151.9–221.7; Table 2).
Statistical summary of log-transformed serum sasanlimab PK parameters following multiple doses of sasanlimab 300 mg Q4W and sasanlimab 600 mg Q6W at steady state (PK parameter analysis set) in phase II.
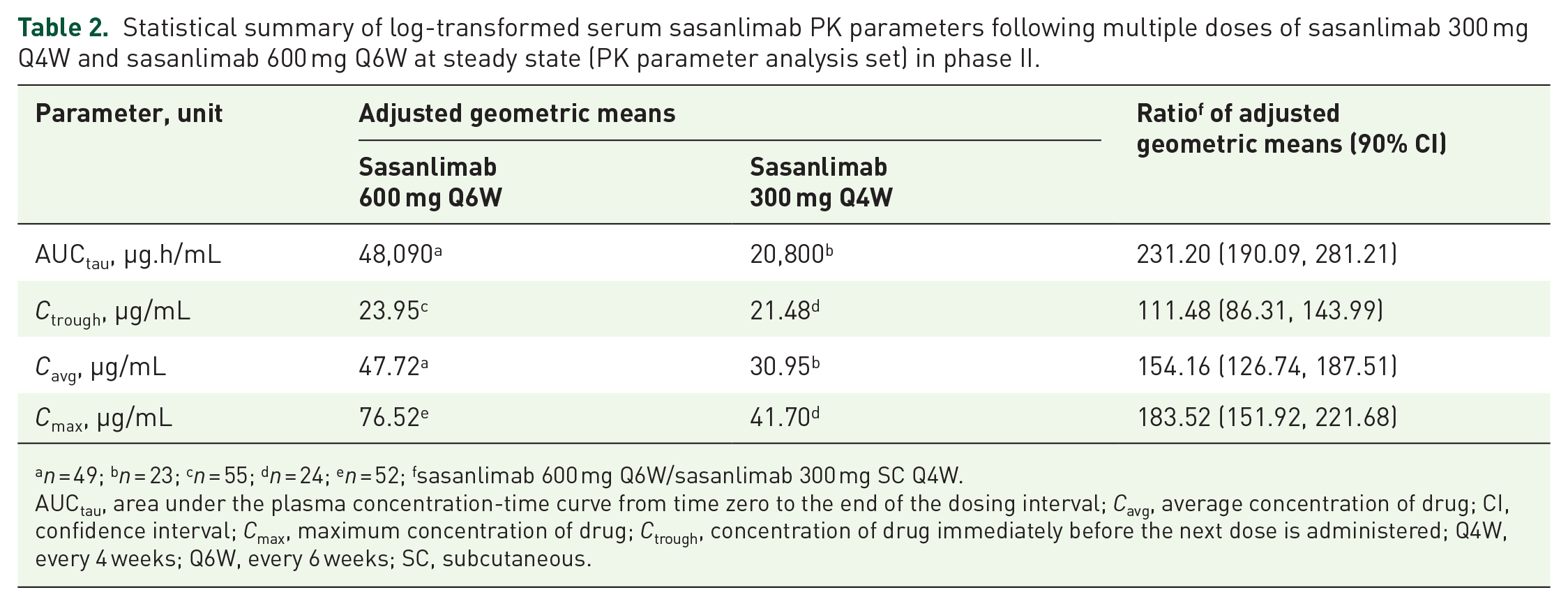
n = 49; bn = 23; cn = 55; dn = 24; en = 52; fsasanlimab 600 mg Q6W/sasanlimab 300 mg SC Q4W.
AUCtau, area under the plasma concentration-time curve from time zero to the end of the dosing interval; Cavg, average concentration of drug; CI, confidence interval; Cmax, maximum concentration of drug; Ctrough, concentration of drug immediately before the next dose is administered; Q4W, every 4 weeks; Q6W, every 6 weeks; SC, subcutaneous.
Immunogenicity
Overall, in phase Ib and phase II, 7 of 142 (4.9%) participants evaluable for ADA analysis experienced a treatment-induced ADA response (300 mg Q4W, 4/52 (7.7%); 600 mg Q6W, 3/90 (3.3%)) and none experienced a treatment-boosted ADA response. Of the ADA events that occurred, no participants had persistent duration of ADA response; 3 of 7 (43%) participants had a transient ADA response, and the remaining 4 of 7 (57%) participants had an indeterminate ADA response. There were no NAbs observed in this study (Supplemental Table 3).
Safety and tolerability profile
Phase Ib
In phase Ib, among 16 participants who received sasanlimab 300 mg Q4W and 18 participants who received sasanlimab 600 mg Q6W, no DLTs were reported in either the dose escalation or dose expansion parts of the study.
In participants treated with 300 mg Q4W in phase Ib, grade 3 TRAEs were reported in 1 of 4 (25%) participants in dose escalation and 3 of 12 (25%) participants in dose expansion, including 2 TRAEs classified as immune-related AEs (irAEs) (immune-related nephritis and immune-related pancreatitis). There were no grade 3 TRAEs or grade 3 irAEs reported in participants treated with 600 mg Q4W in phase Ib and no grade 4 or grade 5 TRAEs or irAEs reported in any dosing regimen (Table 3).
Summary of TRAEs and irAEs.

irAEs, immune-related adverse events; MedDRA, Medical Dictionary for Regulatory Activities; N, total number of participants; Q4W, every 4 weeks; Q6W, every 6 weeks; TRAEs, Treatment-related adverse events.
MedDRA Preferred Terms listed are those reported in 5% or more of participants based on the frequencies observed in any of the dosing regimens of phase II. bAll grade 3 TRAEs reported in any dosing regimens in the study. There were no TRAEs grade 4 or 5 reported.
irAEs by clusters. Each cluster may include one or more MedDRA Preferred Terms. There were no irAEs grade 4 or 5 reported. dFrequency of TRAEs coded to the MedDRA Preferred Term injection site reaction. All injection site reactions were grade 1.
Injection site reactions were reported in one participant in the 300 mg Q4W regimen and one participant in the 600 mg Q6W regimen. Both injection site reactions were grade 1 in severity (Table 3).
Phase II
In phase II, TRAEs were reported in 16/41 (39.0%) participants treated with 300 mg Q4W and 31/80 (38.8%) participants treated with 600 mg Q6W. The most common TRAEs of any grade (⩾5% in any dosing regimen) were pruritis, hypothyroidism, and rash (Table 3). Grade 3 TRAEs were reported in 3/41 (7.3%) participants in the 300 mg Q4W dosing regimen (thrombocytopenia, fatigue, immune-mediated hepatitis, dyspnea) and in 3/80 (3.8%) participants in the 600 mg Q6W dosing regimen (thrombocytopenia, neutropenia, transaminases increased).
The incidence of irAEs was similar in participants treated with the two regimens (Table 3). In the 300 mg Q4W arm, 7 of 41 (17.1%) participants had an irAE of any grade, including 2/41 (4.9%) participants with grade 3 irAEs (immune-related hepatitis, n = 2). In the 600 mg Q6W arm, 13/80 (16.3%) participants had an irAE, including 2/80 (2.5%) participants with a grade 3 irAE (type 1 diabetes mellitus, immune-related hepatitis). The most common irAEs of any grade reported in the sasanlimab 300 mg Q4W and 600 mg Q6W regimens were immune-related rashes (4.9% and 8.8%, respectively), immune-related thyroid disorders (7.3% and 5.0%, respectively), and immune-related hepatitis (7.3% and 1.3%, respectively; Table 3). There were no grade 4 or grade 5 TRAEs or irAEs reported in any of the dosing regimens in phase II.
Injection site reaction in phase II was reported in one (1.3%) participant treated with 600 mg Q6W (grade 1) and was not observed in participants treated with 300 mg Q4W.
Treatment discontinuations
Sasanlimab treatment was discontinued due to TRAEs in 2/12 (16.7%) participants in the phase Ib 300 mg Q4W dose expansion part (pneumonitis and pancreatitis acute) and 1/41 (2.4%) participants in phase II in the 300 mg Q4W arm (immune-mediated hepatitis). There were no discontinuations due to TRAEs in participants treated with 600 mg Q6W in phase Ib or phase II.
Anti-tumor activity
Phase Ib
In phase Ib, one participant with small intestine carcinoma treated with 600 mg Q6W had a CR. A total of 8 participants across dose escalation and expansion had a PR: 4 participants receiving sasanlimab 300 mg Q4W and 2 participants receiving sasanlimab 600 mg Q6W had lung cancer, 1 participant receiving sasanlimab 300 mg Q4W had ovarian cancer and another participant receiving sasanlimab 600 mg Q6W had renal cell carcinoma (Table 4). No OR or SD was achieved in 14 participants (Table 4). Median TTR in the dose escalation or expansion part ranged between 1.8 and 2.8 months for the 2 dosing regimens (Table 4).
Best overall response as assessed by an investigator (full analysis set).

Defined as complete response or partial response.
CI, confidence interval; CR, complete response; N, total number of participants; PD, progressive disease; Q4W, every 4 weeks; Q6W, every 6 weeks; SC, subcutaneous.
Phase II
In phase II, an OR was achieved in 11/41 (26.8% (95% CI, 14.2–42.9)) participants in the sasanlimab 300 mg Q4W arm and 12/80 (15.0% (95% CI, 8.0–24.7)) participants in the 600 mg Q6W arm (Table 4; Supplemental Figure 3). No participants had a CR (Table 4). Median TTR was 2.8 (range: 2.6–11.2) months and 4.2 (range: 2.6–13.8) months in the 300 mg Q4W and 600 mg Q6W arms, respectively (Table 4).
Phase II participants whose tumor was classified as high PD-L1 status (⩾25% of tumor cells exhibit membrane staining) at baseline had higher ORR than participants with low PD-L1 status at baseline in both treatment arms. ORR in participants with high PD-L1 status was 50.0% (n = 12, 95% CI, 21.1–78.9) and 37.5% (n = 16, 95% CI, 15.2–64.6) in the sasanlimab 300 mg Q4W and 600 mg Q6W arms, respectively. In participants with low PD-L1 status, ORR was 15.4% (n = 26, 95% CI, 4.4–34.9) and 8.0% (n = 50, 95% CI, 2.2–19.2) in the 300 mg Q4W and 600 mg Q6W arms, respectively (Supplemental Table 4). Progression-free survival (PFS) was not a prespecified endpoint in the study, but ad hoc analysis of PFS of participants in phase II was conducted, and median PFS was comparable between the 2 treatment arms (300 mg Q4W: 5.6 months (95% CI, 5.3–11.1); 600 mg Q6W: 5.5 months (95% CI, 4.5–6.3); Supplemental Figure 4).
Discussion
In this phase Ib/II trial, the PK, safety, and antitumor activity of sasanlimab 300 mg Q4W and 600 mg Q6W were further explored in Asian participants with advanced malignancies and in global participants with advanced or metastatic NSCLC.
In phase Ib of the study, both dose regimens were shown to have an acceptable safety profile in Asian participants in Japan and China, with no DLTs reported. The PK and safety profiles were generally consistent between phase Ib in Asian participants and phase II in global participants, showing a high degree of overlap. Overall, both regimens were well tolerated and considered safe in Asian participants with no DLTs in phase Ib, similar to previously reported results for sasanlimab. 7
In phase II, the PK analyses indicated that serum sasanlimab exposure based on Cavg and Cmax increased in an approximately dose proportional manner from 300 mg Q4W to 600 mg Q6W. In addition, the ratio for the adjusted geometric mean for AUCtau was 231%, and the geometric mean for Ctrough was 111% steady state for the alternative regimen (600 mg Q6W) compared with the reference (300 mg Q4W) regimen. This demonstrated that exposures for the alternative regimen were not more than 20% lower than the reference regimen, suggesting likely minimal differences in antitumor activity across the two regimens using a PK-based extrapolation approach supported by evidence from the drug class to date. 11 The overall incidence of TRAEs in phase II was similar between participants treated with sasanlimab 300 mg Q4W and 600 mg Q6W. The overall incidence of treatment-emergent ADA responses was low (4.9%), and any differences across regimens were likely due to variability and not considered meaningful.
Despite Cmax exposures that were approximately 84% higher following the 600 mg Q6W regimen, the data showed that the overall safety profile of the 600 mg Q6W regimen in phase Ib and phase II participants was similar to that of 300 mg Q4W with respect to the overall frequencies and severity of TRAEs, including irAEs of any grade or grade 3. The most commonly reported TRAEs and irAEs in both dosing regimens were consistent with those reported in a previous sasanlimab study 7 and demonstrated a similar safety profile to other PD-1 and PD-L1 inhibitors.5,13,14 Further to this, the most commonly observed sasanlimab-related toxicities largely overlap with those reported with other PD-1 and PD-L1 inhibitors (e.g., pembrolizumab, nivolumab, avelumab), which mainly occurred in the skin and endocrine system.14–16 The immune-related AEs were manageable, but some of them led to treatment discontinuation. Injection site reactions were reported in three participants across phase Ib and phase II of the study, all of which were grade 1 in severity, confirming the local tolerability of both dosing regimens.
Antitumor activity, in terms of OR, was observed in phase Ib and phase II of the study at both dosing regimens in participants with advanced solid tumors (phase Ib) and NSCLC (phase II), similar to previously reported results for the sasanlimab first-in-human study 7 and other PD-1/PD-L1 inhibitors.5,13 Though there was a numerical difference in ORR, the confidence intervals overlap implying no substantial differences between dosing regimens for OR in this study. The median PFS in phase II between the dosing regimens were similar. Subgroup analysis of response by PD-L1 status at baseline also suggests a higher response rate for both regimens in participants with high PD-L1 status, in line with published data on other checkpoint inhibitors. 17
A cancer treatment that has a Q6W SC dosing regimen may prove to be advantageous over a 3-week intravenous regimen in some therapeutic settings. SC administration has the advantage of being a less invasive and convenient approach for patients requiring fewer medical resources than intravenous administration and is likely to be associated with fewer days in hospital. 18 Furthermore, SC administration has been reported to be favored over intravenous administration by patients.19–21 A Q6W regimen is also more convenient than a Q3W regimen when combining with conventional platinum doublet therapies and is more convenient as a maintenance therapy as it requires fewer clinic visits. The economic implications of SC administration are also paramount as it reduces healthcare visits (as previously mentioned), administration costs, overall treatment costs, and resource use.5,7,21
Limitations
The open-label design of this study and the different schedule of assessments between the Q4W and Q6W dosing regimens could be considered a limitation as it can introduce potential biases, such as a participant’s reports of side effects and symptoms. While the open-label approach was essential for ethical, practical, and real-world considerations, these biases may impact the study findings. To mitigate these biases, several strategies were implemented such as adhering to standardized protocols, using qualified and trained investigators, and focusing on objective measures. Furthermore, all investigator reports were reviewed to ensure that they adhered to standard guidelines.
Additionally, this study was not designed for a formal comparison of OR, a secondary endpoint; therefore, another limitation of the study is the possible lack of power to objectively demonstrate the difference or lack of thereof in ORR between the two regimens.
Finally, while only Asian participants participated in the phase 1b study, the safety of sasanlimab in a global cohort of patients in phase II indicates sasanlimab to be safe and tolerable at both doses. Moreover, a high degree of overlap was seen between the PK and safety profiles of the phase Ib Asian participants and phase II global participants, indicating a lack of regional or ethnic differences.
Conclusion
In conclusion, this study demonstrated that the sasanlimab 600 mg Q6W regimen achieved similar or higher PK exposures to sasanlimab 300 mg Q4W regimen and showed comparable antitumor activity between the two doses in participants with solid tumors. Commonly observed sasanlimab-related toxicities overlapped largely with those reported with the other PD-1 and PD-L1 inhibitors. Immune-related AEs were manageable, but some led to treatment discontinuation. Overall, both regimens were well tolerated. The comparable benefit–risk profile of sasanlimab 600 mg Q6W versus 300 mg Q4W suggests that it could provide a more practical, convenient, and flexible dosing option for patients with cancer and their physicians. However, further studies are needed in larger and diverse patient populations to help bolster these findings.
Supplemental Material
sj-docx-1-tam-10.1177_17588359241274592 – Supplemental material for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies
Supplemental material, sj-docx-1-tam-10.1177_17588359241274592 for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies by Konstantin Penkov, Igor Bondarenko, Daria Viktorovna Saenko, Yaroslav Kulyaba, Jun Guo, Yi Gong, Noboru Yamamoto, Yevhen Stepanovych Hotko, Vasyl Boyko, Natalya Vladimirovna Fadeeva, Grygorii Mykolaiovych Ursol, Hee Kyung Ahn, Nikolay Viktorovich Kislov, Chia-I Shen, Craig Davis, Karey Kowalski, Elisabete Michelon, Dmitri Pavlov, Tomoko Hirohashi and Byoung Chul Cho in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359241274592 – Supplemental material for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies
Supplemental material, sj-docx-2-tam-10.1177_17588359241274592 for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies by Konstantin Penkov, Igor Bondarenko, Daria Viktorovna Saenko, Yaroslav Kulyaba, Jun Guo, Yi Gong, Noboru Yamamoto, Yevhen Stepanovych Hotko, Vasyl Boyko, Natalya Vladimirovna Fadeeva, Grygorii Mykolaiovych Ursol, Hee Kyung Ahn, Nikolay Viktorovich Kislov, Chia-I Shen, Craig Davis, Karey Kowalski, Elisabete Michelon, Dmitri Pavlov, Tomoko Hirohashi and Byoung Chul Cho in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-3-tam-10.1177_17588359241274592 – Supplemental material for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies
Supplemental material, sj-docx-3-tam-10.1177_17588359241274592 for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies by Konstantin Penkov, Igor Bondarenko, Daria Viktorovna Saenko, Yaroslav Kulyaba, Jun Guo, Yi Gong, Noboru Yamamoto, Yevhen Stepanovych Hotko, Vasyl Boyko, Natalya Vladimirovna Fadeeva, Grygorii Mykolaiovych Ursol, Hee Kyung Ahn, Nikolay Viktorovich Kislov, Chia-I Shen, Craig Davis, Karey Kowalski, Elisabete Michelon, Dmitri Pavlov, Tomoko Hirohashi and Byoung Chul Cho in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-jpg-5-tam-10.1177_17588359241274592 – Supplemental material for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies
Supplemental material, sj-jpg-5-tam-10.1177_17588359241274592 for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies by Konstantin Penkov, Igor Bondarenko, Daria Viktorovna Saenko, Yaroslav Kulyaba, Jun Guo, Yi Gong, Noboru Yamamoto, Yevhen Stepanovych Hotko, Vasyl Boyko, Natalya Vladimirovna Fadeeva, Grygorii Mykolaiovych Ursol, Hee Kyung Ahn, Nikolay Viktorovich Kislov, Chia-I Shen, Craig Davis, Karey Kowalski, Elisabete Michelon, Dmitri Pavlov, Tomoko Hirohashi and Byoung Chul Cho in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-pdf-4-tam-10.1177_17588359241274592 – Supplemental material for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies
Supplemental material, sj-pdf-4-tam-10.1177_17588359241274592 for Pharmacokinetics, safety, and efficacy of an alternative dosing regimen of sasanlimab in participants with advanced NSCLC and other malignancies by Konstantin Penkov, Igor Bondarenko, Daria Viktorovna Saenko, Yaroslav Kulyaba, Jun Guo, Yi Gong, Noboru Yamamoto, Yevhen Stepanovych Hotko, Vasyl Boyko, Natalya Vladimirovna Fadeeva, Grygorii Mykolaiovych Ursol, Hee Kyung Ahn, Nikolay Viktorovich Kislov, Chia-I Shen, Craig Davis, Karey Kowalski, Elisabete Michelon, Dmitri Pavlov, Tomoko Hirohashi and Byoung Chul Cho in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
Pfizer employees designed the study. The principal investigator performed the study. Pfizer employees analyzed the data. All authors contributed to the interpretation of the data, drafting of the manuscript, and critical review for intellectual content. All authors approved the final draft and agreed to be accountable for the manuscript. The authors thank the participants and their families/caregivers, investigators, research nurses, study coordinators, and operations staff who contributed to this study. Medical writing support was provided by Steven Moore, PhD, CMPP, and Haniya Javaid, BSc, of Engage Scientific Solutions and was funded by Pfizer.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.