
Abstract
Background:
Ribociclib has demonstrated a statistically significant overall survival benefit in pre- and postmenopausal patients with hormone receptor positive/human epidermal growth factor receptor 2 negative (HR+/HER2−) advanced breast cancer. New Adjuvant Trial with Ribociclib [LEE011] (NATALEE) is a trial evaluating the efficacy and safety of adjuvant ribociclib plus endocrine therapy (ET) versus ET alone in patients with HR+/HER2− early nonmetastatic breast cancer (EBC).
Methods/design:
NATALEE is a multicenter, randomized, open-label, Phase III trial in patients with HR+/HER2− EBC. Eligible patients include women, regardless of menopausal status, and men aged ⩾18 years. Select patients with stage IIA, stage IIB, or stage III disease (per the anatomic classification in the AJCC Cancer Staging Manual, 8th edition) with an initial diagnosis ⩽18 months prior to randomization are eligible. Patients receiving standard (neo)adjuvant ET are eligible if treatment was initiated ⩽12 months before randomization. Patients undergo 1:1 randomization to ribociclib 400 mg/day (3 weeks on/1 week off) +ET (letrozole 2.5 mg/day or anastrozole 1 mg/day [investigator’s discretion] plus goserelin [men or premenopausal women]) or ET alone. Ribociclib treatment duration is 36 months; ET treatment duration is ⩾60 months. The primary end point is invasive disease-free survival.
Discussion:
The 36-month treatment duration of ribociclib in NATALEE is extended compared with that in other adjuvant cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitor trials and is intended to maximize efficacy due to longer duration of CDK4/6 inhibition. Compared with the 600-mg/day dose used in advanced breast cancer, the reduced ribociclib dose used in NATALEE may improve tolerability while maintaining efficacy. NATALEE includes the broadest population of patients with HR+/HER2− EBC of any Phase III trial currently evaluating adjuvant CDK4/6 inhibitor treatment.
Trial registration:
ClinicalTrials.gov identifier: NCT03701334 (https://clinicaltrials.gov/ct2/show/NCT03701334)
Introduction
The majority (≈80%) of breast cancer cases are nonmetastatic, which is defined as being localized to breast tissue and regional lymphatics. This presentation of the disease is potentially curable with locoregional treatment, such as surgery or radiation therapy, and is also referred to as early breast cancer.1,2 Among patients with early nonmetastatic breast cancer (EBC), the number of patients diagnosed with stage II disease is nearly three times higher than the number of those diagnosed with stage III disease.1,3 In patients with EBC, treatment is administered with curative intent; however, locoregional and/or distant recurrences remain a significant problem in this patient population. Endocrine therapy (ET) is the backbone of adjuvant treatment for hormone receptor positive/human epidermal growth factor receptor 2 negative (HR+/HER2−) breast cancer.4,5 After completion of 5 years of standard ET, considerable risk of recurrence remains in patients initially diagnosed with EBC. Of note, 27–37% and 46–57% of patients with stage II and stage III estrogen receptor positive (ER+) disease, respectively, experience recurrence up to 20 years after diagnosis. 6 Although the risk of recurrence is still highest during the first 5 years after diagnosis, >50% of patients with ER + EBC that recurs experience late recurrences (⩾5 years from diagnosis).6–9 Thus, the prevention of both early and late recurrences are equally important considerations when making adjuvant treatment recommendations for patients with HR+/HER2− EBC. 1
HR +/ HER2− is the most common subtype of breast cancer and accounts for 70–75% of cases. 10 For patients with HR+/HER2− EBC, current European Society for Medical Oncology and National Comprehensive Cancer Network treatment guidelines recommend ⩾5 years of adjuvant ET for most patients after surgery.11,12 Extended treatment with adjuvant ET is recommended for patients with increased risk of recurrence.11,13–15 In addition to ET, treatment with adjuvant chemotherapy may be recommended depending on the risk of recurrence and likelihood of benefit from chemotherapy.
Cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors have shown promising results in the advanced breast cancer (ABC) setting. In particular, ribociclib has demonstrated a statistically significant overall survival (OS) benefit and favorable quality of life (QOL) in both pre- and postmenopausal patients with HR+/HER2− ABC.16–21 Ribociclib has demonstrated a consistent and statistically significant OS benefit in all three Phase III trials of the MONALEESA clinical program for HR+/HER2− ABC.16–18 A noteworthy observation from MONALEESA-2, which evaluated first-line ribociclib, is that patients with de novo metastatic disease experienced a particularly profound OS benefit with ribociclib (hazard ratio, 0.52; 95% CI, 0.36–0.74). 16 Given that patients with de novo advanced disease have not been exposed to prior ET and may be particularly sensitive to treatment, these results are encouraging for investigating the potential efficacy of ribociclib in patients with EBC.
Indeed, the efficacy of CDK4/6 inhibitors in ABC prompted the investigation of these agents in EBC; however, Phase III trials examining adjuvant CDK4/6 inhibitors + ET in patients with HR+/HER2− EBC have shown varying results to date. Palbociclib + ET failed to show invasive disease–free survival (iDFS) benefit in patients with HR+/HER2− EBC in both the PALbociclib Collaborative adjuvant study (PALLAS) and PENELOPE-B trials.22,23 The monarchE trial, which evaluated adjuvant abemaciclib + ET, enrolled patients with four or more positive nodes, or one to three nodes and either tumor size ⩾5 cm, histologic grade 3, or central Ki-67 ⩾ 20%. 24 In monarchE, abemaciclib + ET showed a significant iDFS benefit in the intention-to-treat population at the time of the primary outcome analysis (median follow-up, 19.1 months; hazard ratio, 0.71; 95% CI, 0.58–0.87; nominal p = 0.0009) and in subsequent analyses with additional follow-up.24,25 However, OS data for monarchE are not yet mature, reflecting the chronicity of this disease. While monarchE has shown that abemaciclib reduces the risk of recurrence for up to 4 years and that benefit is sustained beyond treatment discontinuation, it remains unknown whether 2 years of adjuvant abemaciclib treatment is effective in reducing recurrences that could occur even later. Extended follow-up is required to understand the impact of adjuvant CDK4/6 inhibitor treatment on late recurrences and survival.
Additionally, it is important to note that the monarchE trial only included patients with node-positive disease (where N1 also required other high-risk clinical features) and excluded patients with stage II and III N0 disease. As a result, the eligible population comprised only a small portion of the real-world population of patients with HR+/HER2− EBC most commonly seen in the clinic. 26 Given this, a significant unmet need remains for the wider population of patients with HR+/HER2− EBC, including those with N0 disease with additional high-risk clinical or genomic features.
Being a selective CDK4 and CDK6 inhibitor, ribociclib has fewer off-target effects than earlier CDK pan-inhibitors. In addition, preclinical studies demonstrated a greater role for CDK4 versus CDK6 in ER + breast cancer. 27 Ribociclib exhibits preferential binding to CDK4 over CDK6.27–29 Altogether, these data indicate that at clinically relevant doses, including the reduced dose of 400 mg (3 weeks on/1 week off) investigated in EBC, ribociclib may be expected to have the highest on-target inhibition time among the CDK4/6 inhibitor class.30–33
While CDK4/6 inhibitors are known to cause cell cycle arrest, additional evidence suggests that it may have an immune modulatory effect.34,35 Ribociclib has demonstrated an ability to induce an immune environment indicative of a proimmune response in both the preclinical and clinical settings. 34 Additionally, the drug has been shown to induce senescence in cancer cell lines. 36 Altogether, these features of ribociclib may explain its efficacy in ABC for which an OS advantage suggests a carryover effect beyond the treatment period.16,34,36–38 It may be hypothesized that the carryover effect seen with ribociclib in the advanced setting translates into preventing early recurrences, which may be caused by circulating tumor cells, and late recurrences, which may be caused by awakening of dormant cells. 39
The adverse events (AEs) associated with ribociclib are well described, predictable, manageable, and mostly asymptomatic.16,37,38,40 Furthermore, QOL, an important factor driving clinical decisions, has been shown to be maintained or improved with ribociclib plus ET compared with placebo plus ET in ABC.19–21 Compared with the 600-mg starting dose of ribociclib used in the advanced setting, the 400-mg starting dose of ribociclib in the adjuvant setting when tumor burden is lacking is expected to improve tolerability while maintaining efficacy. Additionally, the reduced starting dose of ribociclib in the New Adjuvant Trial with Ribociclib [LEE011] (NATALEE) trial should minimize dose-dependent toxicities that may lead to dose reductions or interruptions; this will likely result in improved adherence and maximized benefits. Overall, the efficacy and safety profile of ribociclib in the advanced setting suggest that it may address the unmet needs of patients with EBC. Thus, NATALEE is evaluating the efficacy and safety of adjuvant ribociclib + ET compared with ET alone in a wide population of patients with HR+/HER2− EBC who have an unmet need.
Methods
NATALEE is a global, Phase III, multicenter, randomized, open-label trial in women, regardless of menopausal status, and men with HR+/HER2− EBC. The NATALEE trial is evaluating the efficacy and safety of ribociclib plus standard adjuvant ET versus standard adjuvant ET alone. Men and premenopausal women also receive a luteinizing hormone–releasing hormone agonist (goserelin). The overall patient population (N = 5101) includes women or men ⩾18 years old. Randomization was stratified by menopausal status, anatomic stage II or III, prior (neo)adjuvant chemotherapy, and geographic region. The enrollment of patients with stage II disease was capped at 40%. A steering committee that comprised investigators, the trial sponsor (Novartis), and Translational Research in Oncology personnel designed the trial and will ensure transparent management of the trial according to the protocol.
Eligibility criteria
Trial eligibility requires completed surgical resection with microscopic margins free of tumor with available archival tumor tissue from surgical specimen or from the diagnostic biopsy for patients who had neoadjuvant therapy and achieved a pathologic complete response. Patients who received prior (neo)adjuvant chemotherapy and adjuvant radiotherapy (if indicated) according to institutional guidelines are required to have completed these treatments >14 days prior to randomization. Eligible patients have a date of initial cytological or histological diagnosis ⩽18 months prior to randomization. Patients are required to have anatomic stage II or stage III disease per anatomic classification in the AJCC Cancer Staging Manual, 8th edition. Enrollment of patients with stage II disease is capped at approximately 2000 patients out of the total trial population. Patients with stage IIA disease may be enrolled if the disease is N1 or N0 or if they have specific features or measures for higher risk of recurrence, such as grade 3 or grade 2 disease. In cases in which the disease is stage IIA or stage N0 or the tumor is grade 2, evidence of high genomic risk is required, such as a Ki-67 score of ⩾20%, an Oncotype DX Breast Recurrence Score of ⩾26, or Prosigna/PAM50, MammaPrint, or EndoPredict EPclin high-risk scores. Patients with stage IIB disease (including N0 and N1) or stage III disease (including N0 and N1) are eligible. Patients with an Eastern Cooperative Oncology Group performance status of 0 or 1 are eligible. Patients are permitted to have already received any standard (neo)adjuvant ET ⩽ 12 months prior to randomization. Key exclusion criteria include prior treatment with a CDK4/6 inhibitor and clinically significant, uncontrolled heart disease, and/or cardiac repolarization abnormalities. Patients are also ineligible if they have distant metastases of breast cancer beyond regional lymph nodes and/or evidence of disease after curative-intent surgery. Patients with prior tamoxifen, raloxifene, or aromatase inhibitor treatment for reduction in risk of breast cancer and/or prior treatment with tamoxifen or raloxifene for osteoporosis within the last 2 years prior to randomization are also excluded. Additional exclusion criteria include major surgery, chemotherapy, or radiotherapy within 14 days prior to randomization and prior treatment with anthracyclines at cumulative doses ⩾450 mg/m2 for doxorubicin or ⩾900 mg/m2 for epirubicin (Table 1).
Key inclusion and exclusion criteria.
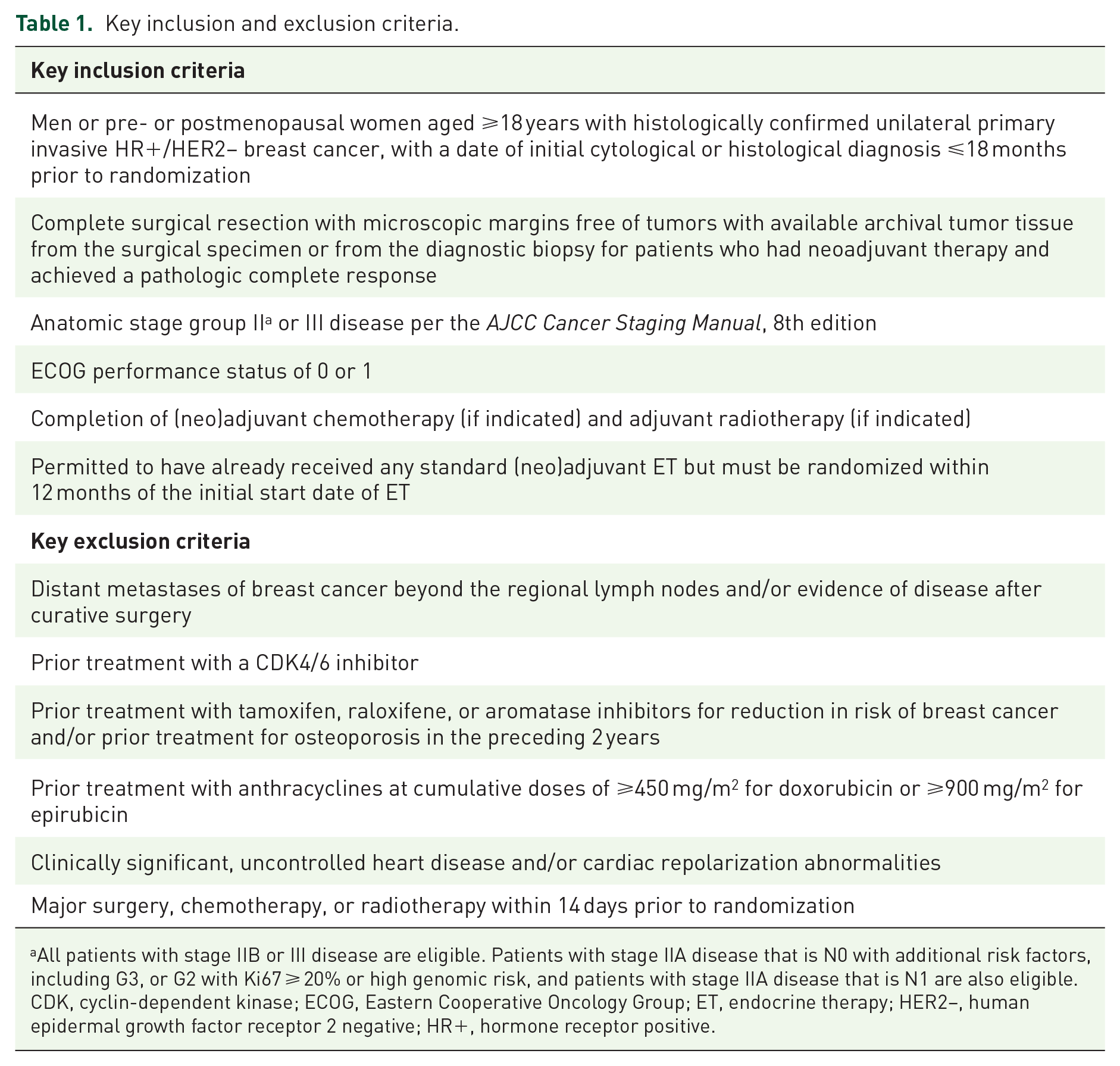
All patients with stage IIB or III disease are eligible. Patients with stage IIA disease that is N0 with additional risk factors, including G3, or G2 with Ki67 ⩾ 20% or high genomic risk, and patients with stage IIA disease that is N1 are also eligible.
CDK, cyclin-dependent kinase; ECOG, Eastern Cooperative Oncology Group; ET, endocrine therapy; HER2−, human epidermal growth factor receptor 2 negative; HR+, hormone receptor positive.
Treatment and scheduling
Patients were randomized 1:1 to receive oral ribociclib in combination with a nonsteroidal aromatase inhibitor (NSAI) versus NSAI alone (Figure 1). Ribociclib is given at 400 mg/day using a 3-week-on 1-week-off schedule. The NSAI partner is either letrozole (2.5 mg/day) or anastrozole (1 mg/day). Choice of NSAI partner is at the discretion of the physician. Men and premenopausal women are also given 3.6 mg of goserelin once every 28 days. Ribociclib is given for 36 months, whereas NSAI treatment (in both arms) is given for ⩾60 months. Patients were required to continue receiving NSAI treatment for the entire 60-month treatment period. In both treatment arms, a change of NSAI was only permitted for protocol-specified reasons, which were the same in both treatment arms. Patients who permanently discontinue NSAI treatment for any reason will proceed to the end-of-treatment and 30-day safety follow-up visits and will remain on trial for the follow-up phase. In the ribociclib arm, a change of NSAI is not allowed, unless it is requested by the patient or if intolerable toxicity or any other medically important event occurs. After the end of treatment with ribociclib and NSAI, investigators are permitted to change ET according to clinical guidelines and local prescription information. Subsequent ET (or any other anticancer treatment) given after the protocol-required 60 months’ duration (or after premature discontinuation of ET in the trial) will be administered according to the investigator’s clinical judgment and is not considered a trial treatment.

Trial design.
Assessments
Trial visits are scheduled to take place at screening, randomization, during treatment, approximately 30 days after discontinuation of ribociclib (investigational arm only), end of treatment (within 15 days after discontinuation of all trial treatments), approximately 30 days after discontinuation of all trial treatments, and during follow-up (Figure 2).

Trial visits and assessments.
End points
The primary end point of the NATALEE trial is iDFS according to Standardized Definitions for Efficacy End Points (STEEP) criteria as assessed by the investigator. The STEEP definition of iDFS includes invasive, ipsilateral breast tumor recurrence, local or regional invasive recurrence, distant recurrence, death (from breast cancer, nonbreast cancer, or unknown cause), invasive, contralateral breast cancer, or second primary invasive cancer (nonbreast cancer). Secondary end points include recurrence-free survival, distant disease-free survival, OS, safety and tolerability, QOL, and pharmacokinetics. Exploratory analyses of biomarkers from tumor tissue or blood samples are also planned. Tumor tissue samples are collected to identify biomarkers that may be predictive of benefit from ribociclib and to assess molecular alterations of genes shown to be associated with HR + breast cancer and the cell cycle. Optional blood samples for pharmacogenetic analysis are collected in consenting patients. Also, in patients who provide consent, optional additional biomarker analyses are planned.
Statistical analysis
Interim analyses are planned after approximately 200, 350, and 425 iDFS events. The final analysis will be after 500 events have occurred. The primary objective of the first interim analysis was to allow for stoppage due to futility with no intent to assess for superior efficacy. The first interim analysis was completed in 2022, and the recommendation was to continue the trial. The second and third interim analyses allow the trial to assess superior efficacy and will only be carried out after all patients have been randomized and (if not withdrawn early) have had at least one postbaseline recurrence assessment. After the final analysis is completed, additional descriptive updates to the efficacy and safety data are planned after 2 years and at the end of the trial, approximately 5 years after the last patient is randomized.
The sample size calculation is based on iDFS. The final analysis will be performed after approximately 500 iDFS events have been documented with approximately 93% power to detect a statistically significant improvement in iDFS with ribociclib + ET versus ET under a target hazard ratio of 0.73.
Participating institutions
NATALEE has included patients from 384 sites in 20 countries: Argentina, Australia, Austria, Belgium, Brazil, Canada, China, France, Germany, Hungary, Ireland, Italy, Poland, Romania, Spain, Taiwan, the Republic of Korea, the Russian Federation, the United Kingdom and the United States.
Discussion
CDK4/6 inhibitors have changed the treatment landscape for patients with HR+/HER2− ABC; however, there are some key differences in the clinical profiles of the three approved agents. OS outcomes for the respective Phase III trials evaluating CDK4/6 inhibitors in the advanced setting highlight some of these differences. All three ribociclib Phase III trials have reported a statistically significant and clinically meaningful OS benefit in pre- and postmenopausal patients.16–18 Ribociclib has set a new benchmark for survival, with an unprecedented median OS of >5 years when combined with letrozole or fulvestrant in the first-line setting in ABC.16,41 While abemaciclib has also shown significant OS benefit in combination with fulvestrant in the second-line setting in MONARCH-2, 42 an interim OS analysis in MONARCH-3 has yet to show a statistically significant benefit of abemaciclib + NSAI versus NSAI alone in the first-line setting; however, interim results suggested a positive trend. 43 Neither Phase III trial evaluating palbociclib in ABC (PALOMA-3 and PALOMA-2) demonstrated a statistically significant improvement in OS.44,45
ET is the current standard of care for patients with HR+/HER2− EBC. Additionally, olaparib has recently been approved for patients with high-risk disease with a germline BRCA1/2 mutation, which occurs in approximately 5% of unselected patients with breast cancer. 46 Currently, there are no data related to treatment sequencing with olaparib and CDK4/6 inhibitors in the adjuvant setting. 12 The addition of CDK4/6 inhibitors to standard-of-care ET in the adjuvant setting has been evaluated in Phase III trials, and notable differences in outcomes were observed among the different CDK4/6 inhibitors. The PALLAS and PENELOPE-B trials both failed to show benefit with palbociclib + ET in the adjuvant setting.22,23,47 To date, only abemaciclib has shown positive results from a Phase III trial for adjuvant treatment of HR+/HER2− breast cancer (monarchE). The monarchE trial included patients who had four or more positive nodes or one to three nodes and tumors that were ⩾5 cm, had a central Ki-67 score of ⩾20%, or were histologically grade 3.24,25,48 Abemaciclib has been approved by the US Food and Drug Administration and the European Commission for use in patients with HR+/HER2− EBC at high risk of recurrence. 25 While National Comprehensive Cancer Network and American Society of Clinical Oncology guidelines have recommended use of adjuvant abemaciclib in the overall high-risk category included in the intention-to-treat population of monarchE, a real-world study indicated that this is likely only ≈10% of the EBC population.10,26,49,50 Given that many patients with stage II disease, who comprise ≈35% of all patients with EBC, still experience recurrence at some point, there remains a significant unmet need for the broader population of patients with stage II and III disease (including N0).1,6
In NATALEE, the risk of recurrence is determined according to stage and nodal status per the anatomic classification in the AJCC Cancer Staging Manual, 8th edition. Patients with stage IIA N0 disease are allowed in NATALEE and required to have other disease features associated with a higher risk of recurrence (grade 3 or grade 2, along with evidence of high genomic risk, such as a Ki-67 score of ⩾20%, an Oncotype DX Breast Recurrence Score of ⩾26, or Prosigna/PAM50, MammaPrint, or EndoPredict EPclin high-risk scores). NATALEE also includes patients with stage IIA disease with one to three positive nodes (N1), any stage IIB disease, and any stage III disease irrespective of nodal status. NATALEE includes node-negative patients and a broader population of patients with stage II and III disease compared with monarchE (Figures 3 and 4); the population in NATALEE is an easily identifiable patient population commonly seen in the real-world setting.1,25 Consequently, results from NATALEE could potentially help address a clinically relevant unmet medical need and may provide an additional option for patients with HR+/HER2− EBC.1,6

NATALEE and monarchE patient populations.
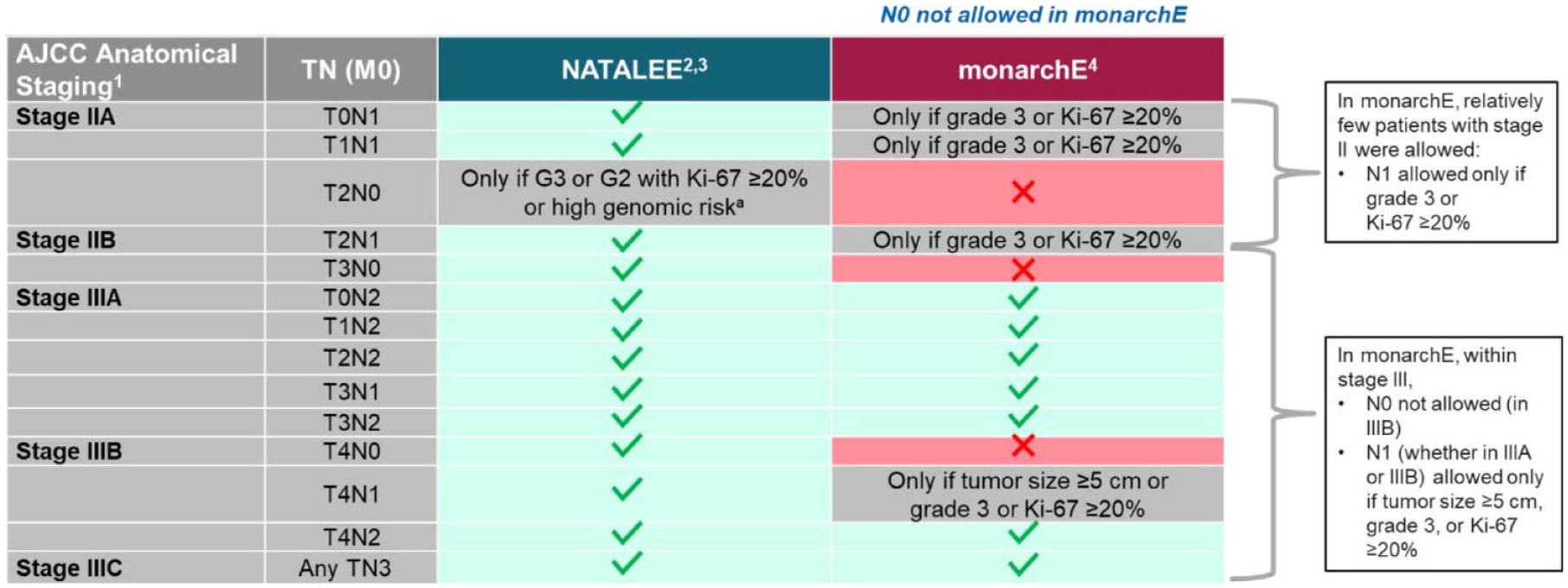
Comparison of NATALEE and monarchE populations.
Recurrence in breast cancer can be driven by multiple and differing mechanisms. Early recurrences may be fueled by replicating circulating tumor cells, whereas late recurrences are likely triggered by awakening of dormant micrometastatic disease.39,51 Longer duration of adjuvant ET has been shown to have an impact on reducing late recurrences – including distant recurrences. 13 It is important to clinically evaluate whether this also applies to CDK4/6 inhibitors. To date, results for abemaciclib have only demonstrated efficacy in reducing early recurrences but have shown that benefit is sustained beyond the 2-year treatment duration (with a most recent follow-up at a median of 4 years). 24 Additional analyses with longer follow-up will be critical in evaluating whether abemaciclib also reduces late recurrences in the high-risk patient population defined by the trial. Prolongation of cell cycle arrest may be important, particularly in the treatment of ER+ disease in which tumor cell dormancy may be long and in which 50% of recurrences occur beyond 5 years. The NATALEE trial is designed to have a 3-year duration of ribociclib treatment in this broader patient population. This is the longest duration of treatment tested among CDK4/6 inhibitors in EBC and was chosen to maximize time-on-target exposure of any circulating tumor cells to ribociclib. This is intended to move these cells from a cell cycle arrest stage to a state of senescence (stable cell cycle arrest), even after ribociclib treatment is stopped. Prolonged cell cycle arrest may decrease recurrences that can occur up to 20 years after diagnosis due to circulating tumor cells.6,7,52–54
Ribociclib, with its unique mechanism of action, has been shown to achieve the highest free drug concentration and selective targeting of CDK4, allowing for increased time on-target for CDK4 inhibition.27,33 Results from preclinical studies suggest that CDK4 inhibition can drive senescence after growth arrest as well as an immune response in cancer cell lines through on-target activity. 36 In an exploratory clinical analysis of immune response using peripheral blood mononuclear cell samples from the RIBECCA trial, ribociclib had significant effects on peripheral innate and adaptive immune responses. 34 Additionally, in the ABC setting, ribociclib demonstrated a carryover effect, resulting in long-term OS benefit,16–18 which may be due to ribociclib’s ability to change tumor biology and induce immunomodulatory effects, impacting response to subsequent therapies. Furthermore, a pooled analysis of the MONALEESA-2, -3, and -7 trials demonstrated a trend toward improved outcomes of subsequent therapies after first-line ribociclib plus ET, suggesting that ribociclib has a posttreatment effect. 55 These unique properties of ribociclib may have the potential to prevent early and late recurrences through direct elimination of replicating tumor cells or by locking dormant cells in a senescent state for immune-mediated clearance, respectively. Based on these results from preclinical and clinical studies, it is hypothesized that ribociclib may induce senescence in micrometastatic tumor cells and an antitumor immune response to ultimately eliminate them.
Symptomatic AEs impact QOL and are also associated with increased nonadherence to treatment, making them critically important clinical considerations in the adjuvant setting when the threshold of patients’ tolerance to AEs is even lower than in patients with ABC.56–59 Abemaciclib is associated with symptomatic AEs, such as gastrointestinal AEs and venous thromboembolic events. In the monarchE trial, patients treated with adjuvant abemaciclib + ET experienced grade 1/2 diarrhea (grade 1, increase of <4 stools per day; grade 2, increase of 4–6 stools per day) and grade ⩾3 diarrhea (increase of ⩾7 stools per day) more frequently than those treated with ET alone (grade 1/2: 75.7% versus 8.5%; grade ⩾3: 7.8% versus 0.2%). 24 Any-grade venous thromboembolic events occurred more frequently with abemaciclib + ET versus ET alone (2.5% versus 0.6%, respectively). 60 Additionally, patient-reported outcome assessments of diarrhea in monarchE favored ET alone over abemaciclib + ET. 60 In the advanced setting, AEs of special interest with ribociclib, such as neutropenia and increased alanine aminotransferase or aspartate aminotransferase, were mostly asymptomatic and identified as laboratory abnormalities.61–63 The 400-mg starting dose of ribociclib in the adjuvant setting, in the absence of tumor burden, is expected to improve the tolerability of ribociclib further by reducing dose-dependent AEs, potentially leading to improved adherence without compromising the intended efficacy. A pooled analysis of the MONALEESA-2, -3, and -7 trials demonstrated that patients who start on 600 mg of ribociclib and require dose reductions for the management of AEs continued to derive progression-free survival benefit with ribociclib.64–66 Additionally, in the three MONALEESA Phase III trials, an OS benefit was maintained among patients with ABC who had a dose reduction from the starting 600-mg dose.65,66 Results from the AMALEE trial in ABC further support that reducing the dose of ribociclib from the starting dose of 600 to 400 mg when needed provides effective management of dose-dependent toxicities without compromising efficacy. 67 Ribociclib showed a favorable safety profile at the 400-mg dose compared with the 600-mg dose, with fewer occurrences of concentration-dependent AEs, such as Fridericia-corrected QT interval prolongation and neutropenia. 67 Ribociclib has demonstrated significant improvements in OS in three separate Phase III studies (MONALEESA-2, -3, and -7) in patients with HR+/HER2− ABC.16–18 In the advanced setting, AEs associated with ribociclib are mostly asymptomatic, and patients reported improved or maintained QOL during treatment.
A recent real-world, multicountry survey of patients and health-care professionals showed that insomnia, diarrhea, back pain, and fatigue had a moderate to severe impact on QOL among patients with ABC who were treated with CDK4/6 inhibitors.68,69 QOL analyses of MONARCH-2 and -3 demonstrated that European Organisation for Research and Treatment of Cancer (EORTC) QLQ-C30 scale scores for diarrhea were significantly worse with abemaciclib compared with placebo.70,71 A matching-adjusted indirect comparison analysis of MONALEESA-2 versus MONARCH-3 demonstrated that time to sustained deterioration of EORTC QLQ-C30 and EORTC-BR23 symptom scales of diarrhea, appetite loss, fatigue, and arm symptoms significantly favored ribociclib over abemaciclib. 72
Patients with recurrent disease have also reported worse QOL compared with patients who remained disease free; therefore, a key goal of adjuvant treatment is to minimize AEs and impact on QOL. 73 It will also be important to understand the economic impact of adjuvant CDK4/6 inhibitor treatment in patients with HR+/HER2− EBC; the duration of treatment with ribociclib in NATALEE is 1 year longer than the recommended duration of adjuvant abemaciclib treatment, whereas the dose of ribociclib used in NATALEE is lower than that used in the ABC setting. The same dose of abemaciclib is used in the treatment of both EBC and ABC. The results of the NATALEE trial will also have important implications for health-care resource utilization, given that economic burden is expected to be higher in the advanced setting compared with the EBC setting, in which curative intent is achieved.74–76
A substantial unmet need for personalized treatment exists in patients with EBC, particularly regarding the use of chemotherapy, given its short- and long-term toxicity implications and intravenous administration. NATALEE does not address whether the addition of ribociclib to standard ET may spare the need for chemotherapy when currently recommended per standard-of-care guidelines. Patients in NATALEE are allowed, but not mandated, to have prior (neo)adjuvant chemotherapy, and it is important to note that prior (neo)adjuvant chemotherapy has been used as a stratification factor in NATALEE. However, other research endeavors with ribociclib are attempting to address this question. 77 Results from the CORALLEEN trial, which compared neoadjuvant ribociclib + ET versus multiagent chemotherapy, showed that patients with high-risk (per PAM50 [Prosigna] risk of relapse) early-stage HR+/HER2− luminal B breast cancer could achieve molecular downstaging of their disease with the combination of ribociclib and ET in a proportion similar to chemotherapy, suggesting that, for some patients, chemotherapy may be avoided if this combination is used. 78 Based on these findings, the RIBOLARIS study (NCT05296746) was designed to evaluate whether chemotherapy could be avoided in patients with clinically high-risk HR+/HER2− disease across the EBC treatment spectrum. 79 Additionally, the ADAPTcycle trial is comparing ET + ribociclib versus chemotherapy in patients with HR+/HER2− EBC at intermediate risk, as defined by an algorithm of clinical and genomic risk factors. 80 In the ABC setting, combination chemotherapy is sometimes used as first-line treatment in patients with aggressive disease characteristics.12,81 The Phase II RIGHT Choice trial investigated whether chemotherapy could be avoided in these patients by comparing ribociclib plus ET with combination chemotherapy in patients with HR+/HER2− ABC and aggressive disease features. 77 The results of RIGHT Choice demonstrated progression-free survival superiority of ribociclib + ET over combination chemotherapy in patients with HR+/HER2− ABC with aggressive clinical features of rapidly progressing or highly symptomatic disease, including visceral crisis. 77 Upcoming data from trials in the EBC setting will be critical to understanding whether CDK4/6 inhibitors plus ET may allow patients to avoid chemotherapy in the adjuvant setting and should provide insight into selecting patients that will benefit from this strategy.
The NATALEE trial is evaluating the broadest population to date of patients with HR+/HER2− EBC and has the longest duration of treatment of any ongoing CDK4/6 inhibitor trial in the early disease setting. Additionally, the reduced starting dose of ribociclib in NATALEE is expected to further minimize dose-dependent toxicities without compromising efficacy. As such, the NATALEE trial has the potential to address many important needs in the treatment of HR+/HER2− EBC that currently remain unmet. NATALEE is currently ongoing, and results are eagerly awaited.
Footnotes
Acknowledgements
We thank the patients who participated in this trial, their families and caregivers, the data-monitoring committee members, the trial steering committee members, and the staff who assisted with the trial at each site. Ribociclib was discovered by Novartis Institutes for BioMedical Research in collaboration with Astex Pharmaceuticals. Medical writing assistance was provided by Casey Nielsen, PhD, of MediTech Media, Ltd, and was funded by Novartis Pharmaceuticals.