
Abstract
Reperfusion injury following ischemic stroke is a complex pathophysiological process involving numerous mechanisms ranging from the release of excitatory amino acids and ion disequilibrium to the induction of apoptosis and necrosis, to oxidative stress and inflammation. The migration of neutrophils into the brain parenchyma and release of their abundant proteases are generally considered the main cause of neuronal cell death and acute reperfusion injury following ischemic stroke. Recent findings in experimental and human stroke have challenged this view, as the majority of neutrophils were rather found to accumulate within the neurovascular unit (NVU) and the subarachnoid space (SAS) where they remain separated from the brain parenchyma by the glia limitans. The brain parenchyma is an immune-privileged site that is not readily accessible to immune cells and does not elicit stereotypic adaptive or innate immune responses. Understanding brain immune privilege requires intimate knowledge of its unique anatomy in which the brain barriers, that include the glia limitans, establish compartments that differ remarkably with regard to their accessibility to the immune system. We here propose that the brain immune privilege also extends to an ischemic insult, where the brain parenchyma does not evoke a rapid infiltration of neutrophils as observed in ischemic events in peripheral organs. Rather, neutrophil accumulation in the NVU and SAS could have a potential impact on cerebrospinal fluid (CSF) drainage from the central nervous system (CNS) and thus on edema formation and reperfusion injury after ischemic stroke. Integrating the anatomical and functional implications of the brain immune privilege with the unquestionable role of neutrophils in reperfusion injury is a prerequisite to exploit appropriate strategies for therapeutic interventions aiming to reduce neuronal cell death after ischemic stroke.
Introduction
A large body of the current literature on neuroinflammatory events following ischemic stroke doesn’t acknowledge that the brain parenchyma is an immune-privileged site not eliciting stereotypic adaptive or innate immune responses as observed in peripheral organs. Understanding brain immune privilege requires intimate knowledge of its unique anatomy, in which endothelial, epithelial and glial barriers establish compartments that differ remarkably with regard to their accessibility to the immune system. We here propose that the brain immune privilege also extends to an ischemic insult, where the brain parenchyma does not evoke a rapid infiltration of neutrophils as observed in ischemic events in peripheral organs. Appropriate consideration of the role of neutrophils in reperfusion injury after ischemia in the brain can thus not rely on observations made in ischemia in peripheral organs. Understanding the anatomical and functional implications of the brain immune privilege will allow exploitation of appropriate therapeutic strategies aimed at reducing neutrophil-induced neuronal cell death after ischemic stroke.
The brain is an immune-privileged organ
Immune privilege is a definition given by immunologists to a tissue site in which experimentally implanted tissue allografts are incapable of provoking adaptive immunity leading to graft rejection. 1 By this definition, the brain is an immune-privileged organ. Subsequent studies have shown that when antigens are introduced into the central nervous system (CNS) parenchyma, they evade systemic immunological recognition irrespective of their bacterial or viral origin. 2 This further supports the notion that the brain parenchyma does not mount a rapid adaptive immune response. Additional observations implicate that the CNS parenchyma similarly lacks a potent innate immune response. Neither injection of bacterial products and thus exposure to pathogen-associated molecular patterns (PAMPs) 3 or injection of chemokines or pro-inflammatory cytokines, 4 nor experimental induction of cell death and thus exposure to damage-associated molecular patterns (DAMPs)5,6 within the CNS parenchyma elicits a rapid infiltration of myelomonocytic cells into the tissue as observed in a stereotypic fashion to such stimuli in peripheral organs.
At the same time, however, the CNS does communicate with the immune system. Tissue grafts are readily rejected when transplanted into the cerebral ventricles. 7 Also, activated T cells can access the cerebrospinal fluid (CSF)-filled ventricular and subarachnoid space (SAS) in the absence of neuroinflammation, 8 supporting the notion that the immune system can access the CNS and that efferent pathways of the immune system to the CNS exist. Furthermore, there is evidence for afferent pathways from the CNS to the peripheral immune system. CSF and interstitial fluid (ISF) constitute the major fluids in the CNS. CSF is an ultrafiltrate of the blood produced by the choroid plexuses (CPs) in all four ventricles of the brain. CSF eventually flows into the SAS where it can then drain into the deep cervical lymph nodes 9 either via pathways crossing the pores of the cribriform plate in the ethmoid bone and lymphatic vessels in the nasal mucosa or via dural lymphatic vessels. 10 In addition, CSF can drain via arachnoid granulations into the venous sinus or along cranial and spinal nerve roots toward lumbar lymph nodes. 9 These pathways have been suggested to also allow for the trafficking of immune cells. 11 ISF is described to be transported along the basement membranes (BMs) of capillaries, arterioles and arteries from the CNS parenchyma to deep cervical lymph nodes. 9 These pathways are, however, too narrow to serve as an exit route for immune cells from the CNS, which may contribute to maintaining immune privilege in the CNS parenchyma. 12 Other studies have suggested a regular mixing of ISF with CSF, mediated by a glial-cell-dependent transparenchymal CSF flow referred to as the ‘glymphatic system.’ 13 While the ‘glymphatic’ concept is difficult to reconcile with the CNS immune privilege, it has recently been suggested that the diffusion-mediated CSF/ISF exchange is rather restricted to the neurovascular unit (NVU) with CSF flow along arterial intramural BMs allowing for ISF/CSF exchange at the level of capillary BMs followed by an outflow of CSF/ISF along BMs of venules and veins, 14 (for review, see Abbott et al. 15 ). This latter concept could be readily reconciled with the observations made on the different accessibility of the immune system to the respective brain compartments underlying CNS immune privilege.
Understanding CNS immune privilege thus requires intimate knowledge of the brain anatomy and its specific compartments that significantly differ with respect to their communication with the innate and adaptive immune system. These compartments are established by the endothelial, epithelial and glial brain barriers. The surface of the brain is ensheathed by three meningeal layers. 16 The outermost layer, the dura mater, is tightly associated with the skull and carries its own blood supply with arteries, veins and lymphatic vessels. The arachnoid mater forms a barrier, the blood–arachnoid barrier, between the dura mater that lacks an endothelial blood–brain barrier (BBB) and the CSF-filled SAS, by forming tight junctions and by the expression of efflux pumps. The SAS carries higher-caliber blood vessels, for example, arteries and veins, descending and ascending from the brain parenchyma, respectively. The avascular pia mater covers the underlying brain and completely surrounds all arteries in the SAS, while coverage of veins remains incomplete. While the pia mater does not provide a barrier for fluids and solutes, it is a barrier for erythrocytes and thus separates the SAS and the parenchymal perivascular compartments as separate entities. 16
The endothelial BBB represents a barrier between the circulation and the CNS compartment. The BBB is composed of a monolayer of highly specialized microvascular endothelial cells (ECs) interconnected by complex and continuous tight junctions. In the underlying endothelial BM, 17 a very high number of embedded pericytes contributes to the unique barrier functions of the BBB endothelium. 18 At the level of postcapillary venules the presence of a second and molecularly distinct parenchymal BM 17 becomes obvious. The parenchymal BM is produced by astroglia that ensheath with their end feet the abluminal aspect of all CNS microvessels. 19 The parenchymal BM and the astrocyte endfeet form the glia limitans superficialis surrounding the entire CNS parenchyma toward the surface, and the glia limitans perivascularis around brain blood vessels. The glia limitans thus ensheath the entire brain parenchyma and form a tight barrier for immune cells in the absence of neuroinflammation. 20 The astrocytes of the glia limitans also establish the structural and functional connection of the blood vessels with the neurons localized in the brain parenchyma. This has led to the recent concept of the NVU referring to the continuous cross talk of the BBB endothelium with the cellular and acellular elements in its direct vicinity.21,22 In its function of maintaining CNS homeostasis, the NVU also strictly controls immune cell emigration from the bloodstream. Immune cell migration into the CNS is strictly controlled by mechanisms that operate at the level of the BBB. During physiological conditions, migration of circulating immune cells into the CNS is very low and limited to specific activated immune cell subsets of the adaptive immune system and probably antigen-presenting cells, which maintain CNS immune surveillance. 12 Immune cells may also enter the CNS via the CPs localized in all brain ventricles. The highly vascularized CPs produce the CSF and lack an endothelial BBB. 23 Circulating immune cells thus have access to the CP stroma where their further passage into the ventricular space is controlled by means of an epithelial cell layer connected by unique tight junctions forming the blood–CSF barrier (BCSFB; for review, see Ghersi-Egea et al. 23 ). In the absence of neuroinflammation, the BBB and BCSFB allow for limited immune-cell access to CSF-drained ventricular and SASs, while the glia limitans prohibit immune cells from entering the CNS parenchyma. Immune privilege of the CNS parenchyma is thus ensured by the brain barriers in an anatomical arrangement resembling that of a medieval castle surrounded by a castle moat, bordered by an outer and inner wall. 24 This unique neuroanatomical setup allows for efficient CNS immune surveillance without disturbing homeostasis of the CNS parenchyma.
Acute events following ischemia in peripheral organs
Recognition of PAMPs by the innate immune system constitutes the host’s first action toward fighting and subsequent elimination of microbial intruders. Additionally, DAMPs are released from necrotic cells enhancing the innate immune response (for review, see Rock et al. 25 ). In case of an ischemic event, the inflammatory process is launched under sterile conditions as opposed to nonsterile inflammation elicited by the body’s exposure to pathogens, for example, bacteria, viruses and fungi. Thus, endogenous DAMPs are the exclusive trigger activating the innate immune system in ischemia.
Expression of pro-inflammatory cytokines and neutrophil chemoattractants
DAMP recognition by their putative pattern recognition receptors either induces directly or precedes an inflammasome-dependent induction of pro-inflammatory cytokines such as interleukin (IL)-1α and IL-1β25,26 by various immune cells, ECs, stromal cells, and platelets. CD11b+ tissue resident macrophages and intravascular CX3CR1+ patrolling monocytes have been described in sensing DAMPs in models of sterile skin inflammation and peritonitis (for review, see McDonald and Kubes 27 ). They immediately associate with the vascular compartment and induce the production of chemokines such as CXCL1 (keratinocyte-derived chemokine, KC) 17 and CXCL2 [macrophage inflammation protein (MIP)-2], 28 central to recruiting neutrophils to the site of ischemic insult.
Neutrophil recruitment across specific sites of the vascular tree
In most organs, postcapillary venules represent the vascular segment for immune cell trafficking across the vascular wall. 29 Immune cell extravasation across the vascular wall in the context of inflammation and ischemia in peripheral organs is a multistep process mediated by the sequential interaction of different adhesion and signaling molecules and their receptors on neutrophils and ECs.30–32 Cytokines such as tumor necrosis factor (TNF)-α and IL-1β induce activation of ECs, which leads to a rapid cell-surface translocation of P-selectin from their intracellular storage, the Weibel–Palade bodies, as well as de novo expression of E-selectin and of the immunoglobulin (Ig) superfamily members intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 on the EC surface.33–35 P- and E-selectin initiate neutrophil interaction with the vascular wall by mediating neutrophil tethering and subsequent rolling by engaging P-selectin glycoprotein ligand-1 (PSGL-1). The reduced speed of the neutrophils allows for detection of inflammatory chemokines such as CXCL1 and CXCL2 bound to the EC surface. Their binding to their cognate G-protein-coupled receptor (GPCR) CXCR2 triggers ‘inside-out’ activation of the β2-integrin lymphocyte function-associated antigen (LFA)-1 (αLβ2; CD11a/CD18) and macrophage-1 antigen (Mac-1) (αMβ2; CD11b/CD18). 36 Activation of integrins leads to their clustering and conformational change from a bent/inactive to a fully extended/active conformation. 37 LFA-1-mediated arrest of neutrophils to endothelial ICAM-1 is followed by Mac-1-facilitated crawling on the endothelium to sites permissive for neutrophil diapedesis. 38 An additional involvement of α4β1-integrin (very late antigen-4) in mediating neutrophil arrest on endothelial VCAM-1 has been proposed by some39,40 but not others 41 as expression of α4β1-integrin on circulating neutrophils is still a matter of debate. 39 Finally, neutrophil diapedesis mainly occurs through the endothelial junctions (paracellular diapedesis) and is mediated by platelet EC adhesion molecule-1, CD99 and junctional adhesion molecule (JAM)-A. 42 Different types of inflammatory stimuli may hereby trigger different mechanisms of neutrophil diapedesis, for example, transcellular diapedesis, which is independent of these junctional molecules. 43
Compartmental localization of neutrophils and interpretation
Assessment of neutrophil contribution to ischemia/reperfusion (I/R) may differ in different tissues and organs depending on the anatomical variations of the respective vascular beds. Thus, lesion pathogenesis in organs with fenestrated endothelium (kidney, liver) may differ from organs possessing a continuous endothelial monolayer (skeletal muscle, brain, mesentery). Presence of neutrophils in tissues after I/R is routinely assessed by isolation of infiltrating immune cells from the affected organ followed by quantitative flow cytometry analysis, which identifies neutrophils by light scatter characteristics combined with positive immunostaining for neutrophil-specific cell-surface antigens (e.g. Ly6G, detected by the antibody clone 1A8 in the mouse and CD15 in man, respectively). A common oversight in the past has been the use of broad-specificity reagents such as the antibody clone RB6-8C5 targeting the Gr-1 antigen, which comprises both Ly6G and Ly6C and therefore does not allow for the discrimination of Ly6Ghigh neutrophils from Ly6Chigh monocytes. 44 While flow cytometry of the immune cell infiltrates allows for precise quantification of the neutrophil contribution in I/R, it lacks any information on their spatial distribution in the tissue. Thus, an immunohistological approach defining the precise localization of neutrophils in the affected organ is desirable. Neutrophils are routinely identified in tissue sections by histological techniques for example, hematoxylin-eosin stain, where their classification as tissue-infiltrating neutrophils relies on recognition of their unique polymorphic nuclei. This histological approach may be hampered by the presence of necrotic or apoptotic cells with pyknotic cell nuclei that could be misinterpreted as neutrophils. 45 Similarly, markers highly suitable to identify circulating neutrophils such as detection of myeloperoxidase may fail in tissue stainings, where upregulated expression of this enzyme in tissue-infiltrating or resident myeloid cells may occur. 46 Thus, combining immunostaining for neutrophil-specific markers, for example, Ly6G and CD45 in the mouse or CD15 and CD45 in man, with antibodies detecting apoptotic cells, for example, activated caspase-3, is essential to unequivocally identify a neutrophil infiltrate in a given tissue.
Acute events following cerebral inflammation
Diapedesis of neutrophils across peripheral continuous endothelial monolayers and their BMs situates the immune cells in the parenchyma of the respective organ irrespective of the inflammatory trigger. There, neutrophils are programmed to eradicate pathogens by releasing oxygen metabolites 47 and proteases 48 or performing Neutrophil Extracellular Trap (NET) activation and release (NETosis), 49 leading to the destruction of bystander cells. In the CNS, the situation is, however, significantly different. Neutrophil migration across inflamed cerebral microvessels or subarachnoid veins, as observed in acute inflammation and meningitis, allows for neutrophil entry into the perivascular or SAS, where they remain separated from the brain parenchyma by the pia mater and the glia limitans. This circumstance is of functional importance, as shown in experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. In this disease model, it has been shown in a number of mouse mutants that even massive accumulation of inflammatory cells in the perivascular or leptomeningeal spaces does not lead to clinical disease. 20 Onset of clinical EAE rather correlates with inflammatory cells breaching the glia limitans and disturbing CNS parenchymal homeostasis.20,50,51 This underscores the important function of this evolutionary old brain barrier as a second barrier for immune cell entry into the CNS parenchyma and thus for maintenance of CNS immune privilege.
Neutrophil contribution to peripheral ischemia
In peripheral vascular beds, P-selectin is stored in Weibel–Palade bodies and, upon an inflammatory stimulus, readily released to the ECs’ surface where it initiates neutrophil tethering and rolling by engaging neutrophil PSGL-1. Subsequent activation of β2-integrins by chemokines presented on the endothelium stabilizes the adhesion of neutrophils via interaction with GPCRs, which results in cell arrest (for review, see McEver 52 ) and subsequent crawling and diapedesis to the side of injury. In the following, we will focus on the role of leukocyte and endothelial CAMs during the cascade of neutrophil recruitment in the peripheral tissue of rodents in response to I/R.
Neutropenia
In a radical approach, the effect of neutropenia was investigated. Thus, reperfusion was conducted exclusively in the absence of neutrophils following myocardial infarction in the dog, resulting in a reduction of both the infarct size and the no-reflow zones. 53 This was interpreted such that neutrophils were indeed mediators of reperfusion injury. This result replicated findings of an earlier study where neutropenia was induced prior to I/R yielding smaller lesions virtually free of neutrophil infiltrate. 54
P-selectin
Neutrophils have been considered the principle therapeutic target affected by anti-adhesive therapies. 55 In a mouse model of myocardial infarction, genetic ablation of P-selectin reduced the lesion area that was thought to be due to reduced neutrophil infiltration observed. 56 However, platelets and leukocytes colocalize in close proximity to ischemic damage. 57 Of note, blocking of P-selectin interferes with platelet function as platelets store P-selectin in α-granules, which upon platelet activation, release P-selectin to the platelet surface where it contributes to platelet aggregation and adhesion to the vascular wall (for review, Furie et al. 58 ). Thus, functional inhibition of P-selectin may affect platelet- or neutrophil-mediated mechanisms of reperfusion injury. On the other hand, there are reports showing that although antibody-mediated blocking of P-selectin in diabetic mice subjected to I/R in the heart reduced neutrophil recruitment, it failed to affect the lesion size. 59 Rather, blocking β2-integrins (CD18-integrins) in the same study reduced both neutrophil recruitment and lesion size. Taken together, these observations suggest that solely reducing the number of tissue-infiltrating neutrophils into the ischemic heart does not suffice for rescuing the tissue. In addition, β2-integrins may play a role in I/R pathogenesis beyond mediating neutrophil extravasation.
In a mouse model of renal failure, genetic ablation or antibody-mediated blocking of P-selectin protected from I/R related tissue damage. 60 The accumulation of platelets along postischemic microvessels via endothelial P-selectin/ PSGL-1 interaction contributed to disease pathogenesis in an intestinal I/R mouse model. 61 Absence of endothelial P-selectin or antibody-mediated blockade could abrogate platelet rolling in the endothelium. Of note, platelet/endothelial interaction was not limited to postcapillary venules but was observed in arterioles as well. 61
P-selectin glycoprotein ligand-1
Antibody-mediated blocking of PSGL-1 decreased reperfusion-induced leukocyte rolling and adhesion in a mouse model of colonic I/R. 62 Application of soluble recombinant PSGL-1 in a cat model of myocardial I/R was able to reduce neutrophil recruitment to the ischemic endothelium. 63
Chemokines
In the ischemic liver, CXC chemokine ligands CXCL-1 and CXCL-2 are induced and deposited on the luminal side of the sinusoids, creating a gradient guiding the neutrophils to the damaged area. 31 CXCL-2 was found to be responsible for initial neutrophil recruitment, 64 and upon inhibition of its respective receptor, curbed neutrophil influx into the liver. 65 Chemokines bind to glucosaminoglycans (GAGs) such as heparan sulfate on the endothelial surface and the extracellular matrix by means of their GAG binding motif. 66 While endothelial heparan sulfate controls chemokine presentation required for the recruitment of lymphocytes to lymph nodes, 67 their role in neutrophil recruitment in an ischemic setting remains to be shown. As a note of caution, the liver vasculature represents very unique features and immune cell/endothelial interactions may deviate in some aspects from the classical paradigm observed in other vascular beds. This includes the absence of selectins in liver sinusoids, the lack of rolling along the endothelium and the fact that integrins are exclusively responsible for neutrophil adhesion (for review 68 ). Adhesion of neutrophils to the liver sinusoids following I/R in the rat is mediated by the interaction of Mac-1 and ICAM-1, and blocking of their function ameliorated the severity of the outcome.69,70
Immunoglobulin-like cell adhesion molecules
In the skin, antibody-mediated blockade of ICAM-1 was demonstrated as protective in a rat epigastric skin flap I/R model 71 leading to flap survival. Another study assessed the contribution of β1-integrins on I/R in a mouse myocardial infarction model. 72 Even though absence of β1-integrins in the hematopoietic compartment reduced the number of infiltrating neutrophils, it did not reduce the lesion size. JAM-C had been shown to mediate neutrophil adhesion to, and diapedesis across, the endothelium following I/R in vitro. Using intravital microscopy in the cremaster muscle model, endothelial JAM-C was observed to redistribute from endothelial junctions to nonjunctional luminal membranes and to mediate neutrophil extravasation after an ischemic insult. 73 Antibody- mediated and pharmacological blocking of JAM-C suppressed leukocyte migration in kidney and cremaster I/R models.
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) have been suggested as playing a role in neutrophil transmigration across endothelial barriers of different vascular beds.74,75 Along that line, application of a phosphinic MMP inhibitor in a liver I/R model in rats resulted in tissue sparing. 76 Interaction of neutrophil α4β1-integrin with the extracellular matrix molecule, fibronectin, was shown to upregulate the expression of MMP-9 accompanied by neutrophil infiltration and expression of pro-inflammatory cytokines. Blockade of that interaction in liver I/R inhibited the process. 77 Neutrophil accumulation in the liver sinusoids does not cause tissue damage (for review, see Jaeschke and Smith 78 ). Only when neutrophils extravasate into the liver parenchyma and interact with hepatocytes, they can degranulate and contribute to lesion pathogenesis. 79
Thus, in peripheral tissues, an ischemic insult is intimately associated with the subsequent tissue infiltration of neutrophils. Nevertheless, reducing their accumulation in the ischemic tissue does not necessarily lead to less tissue injury. These observations point to the existence of additional mechanisms independent of neutrophils, for example, other immune-cell subsets that contribute to I/R injury and tissue damage.
Cerebral ischemia
Approximately 80% of the ischemic strokes in humans involve the middle cerebral artery (MCA) area and thus the model of transient MCA occlusion is very relevant for studying experimental ischemia. In rodent stroke models, transient occlusion of the MCA by the intraluminal filament model leads to damage of the unilateral basal ganglia and adjacent cortical structures, 80 whereas distal occlusion will produce cortical infarction. 81 In either paradigm, neurons are most vulnerable to ischemia and clinical interventions are intended to establish reperfusion (R) to salvage hypoperfused tissue at risk and limit disability in patients suffering from ischemic stroke. However, re-establishing blood flow to formerly non- or hypoperfused areas carries the risk of introducing additional damage by the so-called reperfusion injury. 82 During early timepoints of reperfusion injury, neutrophils are considered to cross the BBB, gain access to the CNS parenchyma and exacerbate tissue injury by releasing their proteases and exerting oxidative stress to surviving neurons in the penumbral region. Experimental stroke models demonstrated that I/R exacerbates the lesion when compared with permanent occlusion. 83 Neutrophils were the first immune-cell subpopulation reported to accumulate in the ischemic brain in rats. 84 Proposing a central role for neutrophils in I/R was supported by clinical findings that showed accumulation of radiolabeled autologous neutrophils adjacent to the lesion in the ischemic human brain following their intravenous re-injection.85,86 Most of the published research has in the meanwhile assigned neutrophils a central role in initiating I/R injury in the brain after ischemic stroke. Neglecting the unique neuroanatomy and the respective role of the brain barriers in maintaining CNS immune privilege has led to the assumption that, after the ischemic insult, neutrophils enter the brain parenchyma where they release inflammatory mediators in apposition to neural cells causing their demise.84,87
To address a causal relationship between the presence of neutrophils and stroke outcome, several studies investigated the outcome of cerebral ischemia following experimentally induced neutropenia. Aside from one report positively correlating neutrophil depletion with stroke volume in a model of transient I/R in the rat, 88 the majority of the studies failed to find a significant beneficial effect of neutrophil depletion on stroke outcome.86,89,90 In the former study, neutropenia reduced the lesion size of the penumbra as opposed to the ischemic core. Thus, the mechanisms by which depletion of neutrophils may be beneficial to stroke outcome need further investigation. If neutrophils were present in the brain parenchyma, a cytoprotective effect of the neurons is conceivable.
An alternative explanation for the beneficial effect of neutrophil ablation relates to their subsequent attraction of further immune cells like monocytes or Th17 cells, which have been shown to contribute to I/R injury at later timepoints after ischemic stroke.91,92 Upon neutrophil depletion in peripheral tissues, Soehnlein and colleagues 93 argue that fewer neutrophils would undergo apoptosis, reducing the release of lysophosphatidylcholine and thus the extent of monocyte recruitment to the site of injury. Consequently, the pro-inflammatory response was blunted and tissue homeostasis enforced (for review, see Soehnlein and Lindbom 94 ).
In addition to experimental depletion of neutrophils, a series of transient ischemia experiments in rodents was performed, aiming to block CAMs potentially involved in neutrophil recruitment to the ischemic brain. Lack of PSGL-1 in mice affected neither the lesion size nor neutrophil recruitment to the CNS when compared with controls following permanent cerebral ischemia. However, when these mice were additionally challenged to develop the autoimmune disease systemic lupus erythematosus, absence of PSGL-1 prevented an increase in lesion size. 95 Antibody-mediated blocking of the αM-integrin subunit of the β2-integrin Mac-1, expressed by myeloid cells, resulted in a reduced lesion size in rats subjected to stroke. 96 However, a clinical trial applying neutralizing antibodies against the αL-integrin subunit of the β2-integrin LFA-1, expressed by all circulating immune cells, failed to translate the experimental findings to the clinic. 97 A role of the endothelial β2-integrin ligand ICAM-1 in neutrophil recruitment across the BBB in stroke was proposed in two studies in ICAM-1-deficient mouse models lacking exon 4 or exon 5 of the ICAM-1 gene, respectively, where absence of ICAM-1 was found to ameliorate the disease.98,99 Again, a subsequent clinical trial failed to translate the beneficial effect of blocking ICAM-1 into the clinic for the treatment of ischemic stroke. 100 EAE studies comparing these different ICAM-1-deficient mice with a novel mouse model lacking the entire ICAM-1 gene 101 subsequently showed that the ICAM-1-deficient mouse models employed in these ischemic stroke studies98,99 still express functional soluble splice variants of ICAM-1 that influence leukocyte function. In a recent study, we found that complete absence of ICAM-1 in ischemic stroke in a mouse model indeed fails to reduce neutrophil accumulation in the CNS, as well as the lesion size. 102
Interestingly, antibody blocking of the α4-integrin subunit in a rat stroke model led to a decreased infarct volume during the acute phase of the disease without reducing the neutrophil recruitment. 103 A multicenter preclinical trial testing the efficacy of blocking α4-integrins in two distinct mouse models of stroke revealed that this treatment significantly reduced both immune cell accumulation and infarct volume after the permanent distal occlusion of the MCA, which causes a small cortical infarction. In contrast, the same treatment failed to show any beneficial effect after transient MCA occlusion, which induces large lesions. 104 This study suggested that benefits of therapeutic targeting of immune cell trafficking may depend on infarct severity and localization. The humanized monoclonal α4-integrin blocking antibody, natalizumab, was recently tested in a placebo-controlled clinical trial (ACTION) in patients with acute ischemic stroke. While natalizumab had no effect on the acute infarct size it reduced the delayed infarct volume when compared with the placebo-controlled individuals. 105 These observations suggest that blocking α4-integrins may reduce monocyte and T-cell recruitment to the ischemic brain rather than blocking acute neurotoxicity mediated by neutrophils and thus affects the progression of neuroinflammation. 106 In the following Phase IIb ACTION 2 study, again performed in individuals with acute ischemic stroke, natalizumab failed, however, to show any improvement in clinical outcomes compared with placebo. As a consequence, further development of natalizumab as a therapeutic regimen in acute ischemic stroke is thus halted (https://www.businesswire.com/news/home/20180207005645/en/Biogen-Reports-Top-Line-Results-Phase-2b-Study).
Experimental antibody-mediated blocking of the α4-integrin ligand VCAM-1 in both, mice and rats did again not interfere with stroke outcome. Of note, monocytes were reduced in number whereas neutrophils remained unaffected after intervention. 107
In conclusion, targeting adhesion molecules in experimental ischemia did not yield homogenous results. If beneficial effects were reported they were mostly limited to experimental stroke and could not be translated into the clinical setting. In conclusion, neither stroke paradigm supports a direct correlation of the presence of neutrophils with the level of neurotoxicity and clinical outcome observed during the acute phase of I/R.
Then, how is one to explain the apparently contradicting findings reported with respect to the role of neutrophils in brain ischemia?
We here propose that most of the studies on cerebral ischemia overlook the fact that neutrophil accumulation in the brain after ischemia does not simply mimic the mechanisms observed in peripheral tissues. In contrast to peripheral vascular beds, ECs of brain parenchymal vessels lack storage of P-selectin in Weibel–Palade bodies. Constitutive expression of P-selectin is limited to meningeal vessels localized in the SAS. Under neuroinflammatory conditions including ischemic stroke, P-selectin expression is induced or upregulated in parenchymal and meningeal venular ECs.45,102,108,109 Delayed availability of P-selectin implies different kinetics of P-selectin-mediated neutrophil recruitment in the CNS. Additionally, as elaborated above, the BBB endothelium strictly controls immune cell entry into the CNS, and the evolutionary older brain barrier, the glia limitans, establishes a second tissue barrier that protects the brain parenchyma from peripheral influences, including immune cell invasion. 110 Ultimately, we suggest that interpretation of the previous studies have failed to consider the presence and function of the brain barriers, which are essential in establishing the immune-privileged status of the brain. The brain barriers establish compartments in the CNS that differ strikingly with respect to their accessibility of innate and adaptive immune cells.
Appropriate consideration of the brain barriers may lead to different interpretations of recent, elegant two-photon imaging studies that have supported the notion of neutrophil infiltration into the CNS parenchyma following ischemic stroke.90,111 A limitation of two-photon imaging is ‘that one only sees what one stains for.’ Thus, the inability to simultaneously visualize the brain barriers and neutrophils after an ischemic insult makes a reliable assignment of the neutrophils to a specific brain compartment challenging, if not impossible. A recent study has, for example, made use of lethally irradiated CX3CR1-eGFP reporter mice to visualize green fluorescent microglial cells reconstituted with bone marrow of CatchupIVM mice that allows for the tracking of red fluorescent neutrophils. By performing two-photon imaging after focal cortical ischemia, the authors observed the extravasation of red fluorescent neutrophils from superficial brain vessels and their subsequent interaction with green fluorescent resident cells of the brain. The observations were interpreted as evidence for neutrophil infiltration of the brain parenchyma where they interact with resident microglial cells. 90 This interpretation did not take into account that CX3CR1 is also expressed by perivascular and leptomeningeal macrophages residing outside of the brain parenchyma.112,113 As these myeloid cells will thus also be visible as green fluorescent cells in the chosen two-photon imaging approach, CX3CR1-eGFP reporter mice do not provide a CNS landmark allowing for the distinction of neutrophil accumulation in the brain parenchyma versus the leptomeningeal space. 90
In fact, we and others have recently observed that following I/R injury, the vast majority of neutrophils accumulate in the leptomeningeal space and the NVU rather than entering the CNS parenchyma following transient and permanent occlusion.45,114 Most importantly, this finding was confirmed in human stroke specimen. Applying antibodies, distinguishing the laminin isoforms present in the endothelial (α4, α5) versus the parenchymal BM 17 (α1, α2) enabled us to define the precise localization of Ly6G+ (mouse) and CD15+ (human) neutrophils within brain tissue sections after ischemic stroke with respect to the brain compartments bordered by these BMs. Based on these observations, the vast majority of neutrophils were found to be localized in the subarachnoid (Figure 1) or perivascular compartments (Figure 2) bordered by the endothelial and parenchymal BMs.45,102,114 These data suggest that even after ischemic stroke, the glia limitans still provided an efficient border prohibiting neutrophil invasion into the brain parenchyma. In addition, we found neutrophils accumulating within arterioles a vascular segment usually not supporting immune cell trafficking (Figure 1).

Accumulation of neutrophils outside of the brain parenchyma following transient cerebral ischemia and reperfusion in the mouse. Neutrophils localize primarily to the subarachnoid space and penetrating arterioles during the acute phase of I/R (a). The font colors of the legend to the cartoon highlight the various cerebral structures involved in I/R. Male C57BL/6 mice were subjected to 45 min ischemia and 24 h of reperfusion. Cryosections were stained with anti-Ly6G (green) to label neutrophils and anti-pan-laminin antibodies to outline the extracellular matrix of blood vessels. There, neutrophils are shown to associate to venous blood vessels [(b), arrow] and distribute in the subarachnoid space during the acute phase of reperfusion [(b), star]. The scale bar is 50 µm.
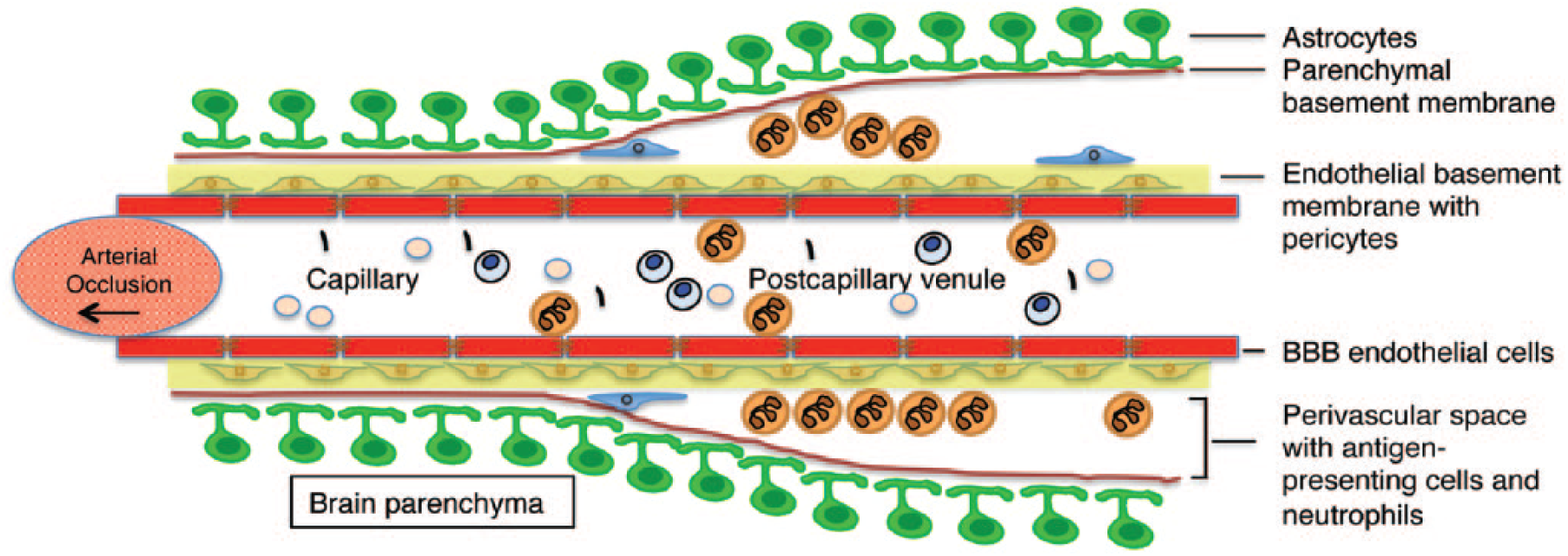
Schematic representation of the vascular and perivascular accumulation of neutrophils in the brain after tMCAO and reperfusion. Neutrophils can cross the endothelial layer of cerebral microvessels but are confined to the perivascular space of postcapillary venules following I/R.
Recent data suggest that neuronal activity and the neurotransmitter glutamate actively relax pericytes and therefore dilate capillaries. 115 When blood flow rises, capillaries dilate prior to arterioles and account for the majority of the increase in blood flow. In the adverse event of ischemia, pericytes around capillaries constrict, eventually leading to pericyte death in rigor and could cause neutrophil trapping in the arterioles. These findings suggest reconsideration of neutrophil involvement in ischemia and reperfusion. Rather than acting neurotoxic, neutrophil accumulation in arterioles may have an impact on the vascular function including CSF drainage recently shown to occur along these pathways.15,116 In fact, cerebral ischemia results in impaired fluid clearance along the perivascular spaces in the affected cortex 117 underscoring a neutrophil-induced malfunction of the NVU in I/R.
Beside their vascular accumulation in cortex-feeding arterioles, neutrophils populate the leptomeningeal space following ischemia. Histologically, neutrophils were detected crossing the endothelial layer of venous blood vessels, dispersing within the confines of the SAS but not entering the brain parenchyma, similar to meningitis. In a rodent model of subarachnoid hemorrhage, neutrophils locate at the SAS and are considered as causing delayed cerebral vasospasms, leading to further restrictions in cerebral blood flow. 118 A similar mode of action of neutrophils detected in the SAS following ischemia is conceivable.
In our studies, presence of neutrophils in the brain parenchyma, that is, beyond the glia limitans, was rare, and often accompanied by extravasated erythrocytes indicating ruptured blood vessels allowing for ‘passive’ release of neutrophils from the circulation rather than providing evidence for their active transmigration across the BBB.
Conclusion
Appropriate consideration of the role of neutrophils in reperfusion injury after ischemia in the brain cannot rely on observations made in ischemia in peripheral organs because the CNS is an immune-privileged organ. While neuronal cell death observed after ischemic stroke is localized to the immune-privileged parenchyma, neutrophils seem to accumulate in the SAS and within the confines of the NVU where they are separated from the brain parenchyma by the glia limitans. In light of the recent observations made on the important role of vascular BMs in CNS blood vessels for CSF and ISF drainage, it is tempting to speculate that accumulation of neutrophils within arterioles, in the perivascular and SAS may have a direct impact on these drainage pathways. Functional impairment of lymphatic drainage from the CNS after ischemic stroke may lead to rapid neuronal cell death due to the accumulation of toxic metabolites in the brain parenchyma.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.