
Abstract
Polymorphonuclear neutrophil granulocytes (PMNs) are part of the early post-ischemic immune response that orchestrates the removal of infarcted brain tissue. PMNs contribute to secondary brain injury in experimental stroke models. In human patients, high PMN-to-lymphocyte ratios in peripheral blood are predictive of poor stroke outcome. Following earlier studies indicating that the cerebral microvasculature forms an efficient barrier that impedes PMN brain entry even under conditions of ischemia, more recent studies combining intravital two-photon microscopy and ex vivo immunohistochemistry unequivocally demonstrated the accumulation of PMNs in the ischemic brain parenchyma. In the meantime, transgenic mouse lines, such as mice expressing Cre-recombinase and the red fluorescent reporter protein tdTomato under the highly granulocyte-specific locus for the gene Ly6G (so-called Catchup mice), have become available that allow study of dynamic interactions of PMNs with brain parenchymal cells. These mice will further help us understand how PMNs promote brain injury and disturb brain remodeling and plasticity.
Introduction
Despite considerable progress in acute stroke treatment, that is, tissue–plasminogen activator-induced thrombolysis and mechanical thrombectomy, where randomized controlled trials (e.g. DAWN, DEFUSE-3, HERMES, WAKE-UP) recently revealed benefits of recanalization treatment that considerably expanded therapeutic windows in acute ischemic stroke,1,2 it remains a leading cause of death and long-term disability. It is well documented that the efficiency and time of vascular recanalization has a strong impact on the severity of ischemic brain injury.1,2 Hence, strategies to reduce the symptom onset-to-recanalization time profoundly affect the stroke outcome. 3 In contrast, approaches aiming at promoting neuronal survival in the first hours by neuroprotectants have lost priority in the stroke field, given that neuroprotectants repeatedly failed in clinical trials. 4 Ischemic stroke is followed by a plethora of secondary events that exacerbate brain injury and promote neurodegeneration over several days to weeks poststroke. 5 Considering the fact that, despite optimized recanalization therapy, most stroke patients still exhibit neurological deficits in the postacute stroke phase, there is still an urgent need for restorative treatments that promote brain remodeling, plasticity and repair.
Postischemic neuroinflammation comprises the early (within minutes to hours poststroke) infiltration of the injured brain by polymorphonuclear neutrophils (PMNs) and monocytes/macrophages.5–7 This is accompanied by the activation of microglia and expression of proinflammatory cytokines, adhesion molecules and integrins, which are supposed to contribute to ischemic reperfusion injury.5,8,9 Massive infiltration of PMNs into ischemic brain tissue has recently been documented in human stroke patients. 10 In peripheral blood, high PMN-to-lymphocyte ratios (>4.8) before initiation of thrombolysis predicted poor outcome 3 months poststroke, 11 supporting a detrimental role of PMNs in reperfusion injury and brain remodeling. Considering their early brain accumulation and central role in coordinating subsequent immune responses, PMNs might represent pivotal targets which profoundly modify recovery processes. In this study, we aim to summarize existing evidence regarding the roles of PMNs in the ischemic brain, defining how cell-type-specific immunodepletion and fluorescence microscopy studies have recently contributed to understanding how PMNs influence brain injury and neurological recovery.
Role of polymorphonuclear neutrophils for ischemic reperfusion damage
PMNs have been shown to be part of the early inflammatory response after focal cerebral ischemia several years ago.12,13 The prevention of PMN brain entry via blockade of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) was found to confer neuroprotection in animal models of ischemic stroke,14,15 suggesting a causative role of PMNs in the development of reperfusion injury. Detection of PMNs has been suggested to be confounded by the cross-reaction of antibodies with epitopes of monocytes/macrophages. As such, a frequently used antibody targeting the Gr-1 antigen recognizes both PMN Ly6G and monocytic Ly6C, and hence lacks PMN specificity. This is also true for myeloperoxidase staining, which detects PMNs, monocytes and microglial cells. 16 Furthermore, inhibitors of PMN brain entry in these early times were not specific for PMNs, but also inhibited other cells, such as monocytes or macrophages. 17 Perhaps, as a consequence of lacking immune-cell specificity or wrong timing of immune-cell inhibitor delivery that may have been initiated too late, early clinical trials preventing PMN brain entry (e.g. Enlimomab, ASTIN 18 ) did not find improvements of neurological recovery in human stroke patients.18,19
Aiming to define the role of PMNs in the development of ischemic reperfusion injury, Enzmann and colleagues exposed mice to focal cerebral ischemia using the transient intraluminal middle cerebral artery occlusion (MCAO) technique, distinguishing infiltrating PMNs from monocytes/macrophages by cell-type-specific antibodies (Ly6G and Ly6C, respectively). 20 In their study, the authors did not find any significant numbers of PMNs in infarcted brain parenchyma, but only trapped in perivascular spaces or the luminal side of cerebral blood vessels. 20 Following similar observations in infarcted brain tissue of human stroke patients at different timepoints poststroke, the authors concluded that PMNs do not invade ischemic brain to any significant extent. However, this view has been challenged by a number of other studies5,6,10,21 and later, also, a partly revised view of the original authors appeared. 7
Important roles in ischemic reperfusion injury have been demonstrated for peripheral T cells, although the exact mechanisms remain unclear, and the main influx of T cells occurs much later than that of PMNs.6,22 In a combination of studies using lymphocyte-deficient Rag2−/− mice and wildtype mice, in which CD4+ and CD8+ T cells had been depleted by anti-CD4 and anti-CD8 antibody, respectively, Liesz and colleagues had shown that invading T cells contribute to ischemic injury both via humoral secretion (e.g. release of interferon-γ) and cytotoxic (i.e. expression of perforin) mechanisms. Since depletion of mononuclear/myeloid cells by anti-Gr-1 antibody did not have any major effect on reperfusion damage, these experiments were interpreted in favor of T cells but not PMNs as contributors to reperfusion damage. 6
Using intravital two-photon microscopy combined with conventional immunohistochemistry, we have subsequently shown after permanent distal or transient intraluminal MCAO in mice that PMNs rapidly attach to inflamed cerebral endothelium followed by transmigration of cells into the brain parenchyma. 5 Invading PMNs rapidly interacted with local microglia and were phagocytosed by them,5,23 a behavior previously observed in tissue-based ischemia models in vitro. 8 Unlike in earlier studies, PMNs were unequivocally found to contribute to ischemic brain injury: PMN depletion using an antibody directed specifically against the surface protein Ly6G potently reduced ischemic injury in mice, in which CD3+ T cells had been depleted, as did blockade of PMN brain entry by an anti-very-late-antigen-4 (VLA-4; i.e. α4-integrin) antibody. 5
Importantly, there was a clear synergistic effect of CD3 and VLA-4 blockade in protecting mice from the behavioral deficits associated with experimental stroke, while such a synergism was lacking in the combination of anti-Gr-1 or anti-Ly6G antibodies with anti-VLA-4. Hence, VLA-4-mediated PMN entry into brain was a critical inducing factor of early behavioral deficits, while T-cell entry was not. 5 VLA-4-mediated PMN brain entry was unexpected, since VLA-4 had previously been shown to mediate T-cell brain entry. 24 Meanwhile VLA-4-mediated PMN brain entry has been confirmed by other groups, 7 and brain-invading PMNs have been shown to form extracellular DNA traps 10 that are supposed to damage central nervous system neurons directly 25 or via induction of coagulation processes.7,10 PMNs closely interact with platelets in the ischemic microvasculature, 13 mutually activating each other, resulting in impaired reperfusion as a consequence of platelet-induced microthrombosis and brain hemorrhage as a consequence of PMN-induced extracellular matrix breakdown. 26 PMN infiltration to the brain was found to be platelet dependent, and was reversed by blockade of the platelet receptor glycoprotein Ibα. 27 The role of PMNs for stroke outcome has recently also been confirmed in an elegant transgenic animal model. Here, it was shown that PMN-expressed CD39 (a cell-surface adenosine triphosphatase) inhibits PMN invasion into the brain and hence leads to significantly reduced behavioral deficits in experimental stroke. 28
PMN depletion by anti-Ly6G antibody and blockade of PMN brain entry by anti-VLA-4 antibody consistently improved motor coordination deficits in mice exposed to transient intraluminal MCAO, as evaluated by a battery of motor coordination tests. 5 In contrast, T-cell depletion did not induce such motor coordination changes in the same study, despite reduction of brain infarct volume. 5 These latter data suggest that PMNs might have particularly pronounced deleterious effects on brain remodeling and plasticity, in addition to the exacerbation of ischemic brain damage.
The Effect of Natalizumab on Infarct Volume in Acute Ischemic Stroke (ACTION) trial
Based on these and other findings, a randomized, double blind phase II multicenter study, ACTION, was conducted and recently reported functional benefits of human stroke patients in 161 patients aged 18–85 years assigned to the VLA-4 inhibitor natalizumab (300 mg, intravenously) or placebo within 9 h after ischemic stroke onset.29,30 Natalizumab, which is clinically used in multiple sclerosis patients because of its specificity of VLA-4 actions that profoundly impede leukocyte entry into the inflamed brain, did not influence the primary endpoint of the study, that is, infarct volume growth from baseline to day 5 compared with placebo [median absolute growth 28 ml (range −8 to 303 ml) versus 22 ml (−11 to 328 ml); relative growth ratio 1.09 (90% CI 0.91–1.30), p = 0.78] and did not change the National Institutes of Health Stroke Scale (NIHSS) score at various time points. Yet, more patients in the natalizumab group than in the placebo group had a modified Rankin scale (mRS) score of 0 or 1 (indicative of little or no functional neurological deficits) at day 30 [13 (18%) versus 7 (9%); odds ratio 2.88 (90% CI 1.20–6.93), p = 0.024] and day 90 [18 (25%) versus 16 (21%); 1.48 (0.74–2.98), p = 0.18], or a Barthel index score ⩾ 95 (indicative of functional independence) at day 90 [34 (44%) versus 26 (33%); 1.91 (1.07–3.41), p = 0.033], which were secondary endpoints of this study. Natalizumab and placebo groups had similar incidence rates of adverse events, serious adverse events and deaths. 30
The ACTION trial concluded that a phase III multicenter trial should be conducted in view of functional benefits induced by VLA-4 blockade. However, another very recent phase II study, ACTION-2, failed to show beneficial effects of natalizumab in approximately 270 acute stroke patients (http://media.biogen.com/news-releases/news-release-details/biogen-reports-top-line-results-phase-2b-study-natalizumab-acute). This has stopped the manufacturer of the drug from pursuing further phase II trial activities using natalizumab for stroke treatment. However, as long as more detailed data on this second trial are not available, it is difficult to ponder potential reasons for the lack of clinical efficacy. As a matter of fact, the lack of clinical efficacy may be a simple consequence of the lack of statistical power in both rather small phase II trials. Apparently, the ability of natalizumab in preventing PMN brain entry could not be determined in both ACTION trials. Importantly, only a subset of human PMNs express VLA-4 31 and even simple brain-affecting measures such as sleep deprivation have been shown to reduce the expression level and number of VLA-4-expressing PMNs in humans. 32 Hence, it might be premature to surmise on these negative findings.
Role of polymorphonuclear neutrophils in ischemic injury associated with hyperlipidemia and hyperglycemia
Compared with normolipidemic wildtype mice, hyperlipidemic apolipoprotein-E−/− (ApoE−/−) mice develop particularly severe brain injury in response to focal cerebral ischemia, which is associated with elevated granulocyte counts in spleen and blood. 33 Importantly, studies in ApoE−/− mice revealed that Ly6G antibody-induced PMN depletion or inhibition of PMN brain entry by pharmacological CXC motif receptor-2 (CXCR2) blockade or delivery of a neutralizing CXCR2 antibody abolished the augmentation of ischemic brain injury induced by hyperlipidemia, indicating that PMNs are responsible for the exacerbated brain injury attributed to this stroke risk factor. 21 In hyperglycemia, similar observations, that is, exacerbated brain PMN inflammation associated with increased brain injury, were made. 26 So far, the molecular mechanisms by which invading PMNs compromise brain remodeling are unknown. It has previously been shown that PMNs are not harmful to neurons in healthy hippocampal brain slices but adopt a deleterious phenotype that destroys neurons when invading ischemic brain slices. 8 Thus, ischemic microenvironments appear to trigger mechanisms in PMNs which augment brain damage and prevent restorative processes.
Transgenic Ly6GtdTomato mouse allows evaluating role of polymorphonuclear neutrophils in ischemic brain
Until recently, the investigation of PMN function in mice was hampered by the lack of animal models that would allow the cell-type-specific visualization and manipulation. Previously available models such as the LysEGFP mouse 34 that we used in our intravital studies were helpful for imaging but suffered from the fact that not all enhanced green fluorescent protein (EGFP)-expressing cells were PMNs. 5 The same is true for mice expressing Cre-recombinase from the LysM locus. 35 Also here, deletion of genes is not specific for PMNs. A recent study has tested all mouse lines available at the time for their specificity regarding PMN action and found none to be totally convincing. 36
In light of this fact, a novel mouse model has been generated, in which the highly PMN-specific locus for the gene Ly6G was genetically manipulated to drive the expression of both, Cre-recombinase and the red fluorescent protein tdTomato. This novel mouse line has been termed ‘Catchup’ and is superior in PMN specificity to any other animal model available so far. 37 Next to the PMN-specific deletion of conditional alleles in vivo, it also allows the intravital imaging of red fluorescent PMNs.23,37–42 Importantly, PMNs in this mouse are functionally entirely normal and also present in physiological numbers. This Catchup mouse line can successfully be crossed to mice with a floxed allele of alpha-4 integrin 43 which leads to a PMN-specific deletion of VLA-4 without affecting T cells. Otherwise, the mice are phenotypically normal. In the future, this mouse line will allow testing the impact of VLA-4 as entry receptor for PMN to ischemic brains but also any other molecule found associated with brain infiltration of PMN in a cell-selective manner.
A prerequisite for studying the molecular function of PMNs in inflamed tissues is the ability to quickly isolate the cells at high purity. Any sorting approach using direct cell labeling (e.g. by antibodies) is problematic, as antibody binding severely influences PMN function. 44 The Catchup mouse offers the unique possibility of isolating highly pure PMNs in a simple one-step sorting procedure based on their endogenous red fluorescence without additional labeling (i.e. the PMN remain ‘untouched’). Using this approach is successful if we have managed to perform isolations of pure PMNs from bone marrow and solid tissues in a fast one-step sorting approach that can be used for studying dynamic responses of PMNs to ischemic brain injury.
In order to measure and quantify the interaction of PMNs with other mononuclear cells in vivo, it is essential to visualize two cell types simultaneously and in different colors. By crossing Catchup mice to mice with microglia-specific EGFP expression (CX3CR1EGFP mice), 45 it has become possible to generate mice that allow studying interactions of PMNs and microglia [Figure 1(a)]. Importantly, these mice do not exhibit any overlap of EGFP and tdTomato expression. 46 Instead, analysis of circulating leukocytes by flow cytometry shows perfectly separated EGFP and tdTomato-expressing cells [Figure 1(b)]. Hence, green and red cells are clearly separated, thereby allowing the simultaneous observation of both PMNs and microglial cells [Figure 1(c), (d)]. Furthermore, in the brains of CX3CR1EGFP mice reconstituted with bone marrow from CatchupIVM mice, we detected clearly separated PMN and microglia signals, including their physical contacts after stroke (unpublished).
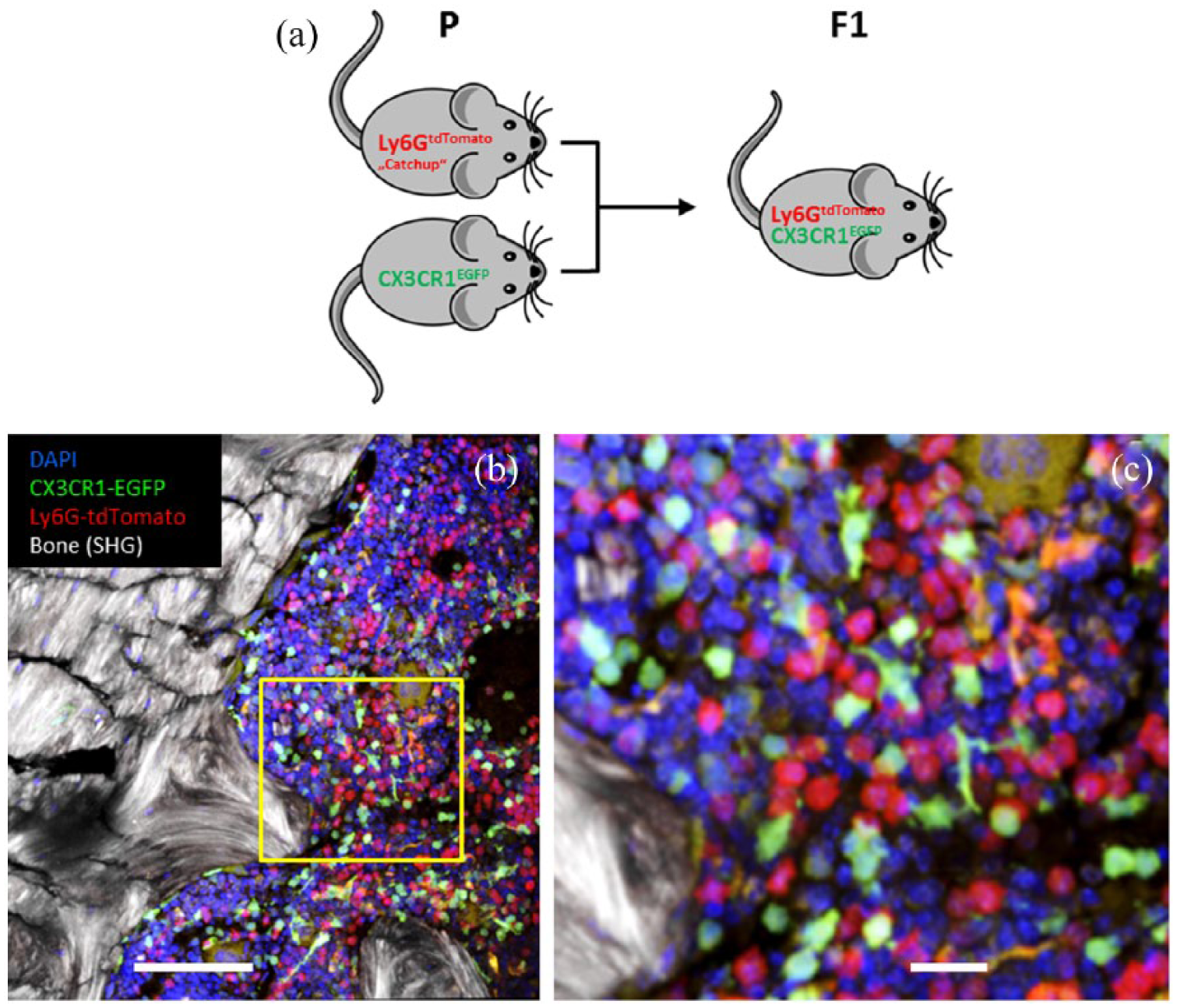
CX3CR1EGFP mice with microglia-specific EGFP expression crossed with Catchup mice show selective expression of tdTomato in polymorphonuclear neutrophils and EGFP staining in macrophages, dendritic cells and microglia.
Next to visualizing the dynamics of cellular interactions by intravital microscopy, it will be centrally important to reliably study restorative processes of the ischemic brain, where microvessels and parenchymal cells (specifically, neurons) exhibit profound remodeling.47–49 Since the inhibition of PMN brain entry reduces ischemic injury, at the same time enhancing neurological recovery, 5 the enhanced recovery should go along with better preservation of microvessels, reduced vascular remodeling, reduced neurodegeneration and improved neuronal plasticity.
In an initial attempt to understand the pathophysiological role of PMN in stroke, we investigated the production of neutrophil extracellular traps (NETs), which are DNA strands decorated with citrullinated histone-H3 (H3-Cit) that are released by PMNs under pathophysiological conditions. 50 NETs have previously been reported in brains of patients and animal models of Alzheimer’s disease 51 and stroke, 10 and NET-associated proteins are known to be neurotoxic. 25 Indeed, we found ample NET production in stroke-affected brain areas with infiltrated PMNs (Figure 2). We hypothesize that NET production contributes to PMN-induced ischemic brain damage. These experimental systems and working hypotheses will form the basis of future work on clarifying the role of PMNs for the exacerbation of stroke.

Neutrophil extracellular trap production in stroke-affected murine brains.
Conclusion
From a large number of experimental studies, there is ample evidence for a fundamental, mostly detrimental role of PMNs in the ischemic brain. Unfortunately, so far this observation could not be translated into novel treatment options for human stroke patients. Future research should more thoroughly characterize this important inflammatory cell type to determine the Achilles heel of their function that can be successfully targeted in the clinical setting. Such interventions should, however, be evaluated with caution, since physiological functioning of peripheral PMNs is essential for immune defense in stroke patients.
Footnotes
Acknowledgements
Funding
This research was supported by The Deutsche Forschungsgemeinschaft (HE3173/11-1, GU769/10-1).
Conflict of interest statement
The authors declare that there is no conflict of interest.