
Abstract
Chronic hepatitis C virus (HCV) infection is a leading cause of chronic liver diseases and hepatocellular carcinoma (HCC) worldwide. In the past few years, anti-HCV therapies have undergone a revolution with the approval of multiple direct-acting antivirals (DAAs), which enable interferon-free treatments with considerable improvement of sustained virologic response in patients. Today, DAAs have become the standard of care for HCV therapy. However, several limitations remain, which include access to therapy, treatment failure in a subset of patients and persistent risk of HCC development following cure in patients with advanced fibrosis. By targeting conserved host proteins involved in the HCV life cycle, host-targeting agents (HTAs) offer opportunities for pan-genotypic antiviral approaches with a high barrier to drug resistance. Moreover, when applied in combination with DAAs, HTAs could improve the management of difficult-to-treat patients by acting through a complementary mechanism of action. In this review, we summarize the different HTAs evaluated in preclinical and clinical development and discuss their potential role for anti-HCV therapies.
Introduction
Hepatitis C virus (HCV) is a hepatotropic RNA virus, which belongs to the Flaviviridae family. 1 Chronic HCV infection is a major public health problem, affecting more than 150 million individuals worldwide. Chronically infected patients are at high risk for developing liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC). In many parts of the world, HCV infection is the major cause of HCC and the leading indication for liver transplantation (LT). 2 There is no vaccine to prevent HCV infection. In the past, interferon (IFN)-based regimens were the standard of care for HCV infection, but only led to a sustained virologic response (SVR) in 50% of patients with serious adverse effects. 3 Recent approval of novel antivirals directly targeting the virus, named direct-acting antivirals (DAAs), have enabled IFN-free treatments with considerable SVR improvement (SVR rates over 90%). Although the development of DAAs has revolutionized HCV therapy, several limitations remain: these include limited access to therapy in the majority of infected patients, treatment failure in a subset of patients, potential adverse effects in patients with comorbidity and persistent HCC risk following SVR in patients with advanced fibrosis. 4 Targeting host factors required for virus infection is an attractive complementary strategy to address these challenges. An improved understanding of the viral life cycle based on the development of advanced HCV model systems has enabled the design of new molecules that target key factors of the HCV life cycle, named host-targeting agents (HTAs). 5 HTAs provide a broad antiviral activity with very high genetic barrier to drug resistance due to the extremely low mutational rate occurring within host cells.5,6 Several HTAs are now being evaluated in phase II and III clinical trials. Here, we review the different classes of HTAs in preclinical or clinical development and highlight their future role in anti-HCV therapy.
Treatment of HCV infection in the era of DAAs
In recent years, the treatment of chronic HCV infection has dramatically improved with the development of IFN-free regimens based on DAAs. Indeed, a better understanding of the HCV life cycle has led to the development of multiple DAAs, with highly improved SVR rates, shortened treatment duration and reduced side effects. 7 DAAs are molecules that specifically target defined nonstructural (NS) viral proteins playing a crucial role in the HCV life cycle. At least four classes of DAAs are available in the US and Europe: NS3/NS4A protease inhibitors (e.g. simeprevir, grazoprevir, paritaprevir), NS5B nucleoside and non-nucleoside polymerase inhibitors (e.g. sofosbuvir and dasabuvir, respectively) as well as NS5A inhibitors (e.g. daclatasvir, ledipasvir, ombitasvir). 3 Combinations of DAAs are currently the standard of care for patients with HCV infection. In 2014, the combination of sofosbuvir and ledipasvir (Harvoni, Gilead, Foster City, CA, USA) was approved for the treatment of HCV genotype 1 infection, with a SVR of more than 95%.8–10 Moreover, in the same year, the US Food and Drug Administration (FDA) also approved the combination of three DAAs, namely ombitasvir, paritaprevir and dasabuvir (Viekira Pak, Abbvie, North Chicago, IL, USA) for the treatment of HCV genotype 1 infection.11–13 In 2016, additional DAA-based regimens including sofosbuvir/velpatasvir (Epclusa, Gilead) and grazoprevir/elbasvir (Zepatier, Merck, Kenilworth, NJ, USA) were approved for the treatment of pan-genotypic HCV infection with a SVR rate of about 95%.14,15
Limitations of DAA-based therapies
Today, it is estimated that more than 90% of patients with chronic hepatitis C can be cured with DAA-based regimens. Clinical studies involving large numbers of patients confirmed excellent efficacy, safety and tolerability of the new DAA combinations. However, several challenges remain unsolved.
The most important limitation is probably the accessibility of DAA regimens. Indeed, access to DAAs is limited to less than 10% of patients with HCV infection, especially in low-resource countries. 16 Moreover, the management of special populations, or ‘difficult-to-treat’ patients still requires special attention. 17 One challenge remains the treatment of patients with advanced cirrhosis and decompensated liver disease. Recent studies revealed that patients with or without cirrhosis respond equally well to DAAs, whereas patients with advanced cirrhosis appear to have a reduced ability to clear the virus, leading to lower SVR rates in this population.18,19 For these patients, treatment regimens should be adapted to higher doses or longer treatment duration. In this regard, it is important to note that patients with advanced cirrhosis (Child-Pugh classes B and C) were excluded in many phase II and phase III clinical trials and fewer studies were conducted in patients with decompensated liver disease in the past.8,10–12,20–22 Consequently, the dosage of DAAs, either alone or in combination, their efficacy and their safety have only been partially addressed in these special populations. Furthermore, several DAA-based regimens (e.g. the NS34A protease inhibitors simeprevir and asunaprevir) cannot easily be used in patients with decompensated cirrhosis because of their impaired drug-metabolizing capacity, leading to severe adverse effects.19,23,24 DAA-based regimens can also be challenging for patients with advanced renal insufficiency who often need DAA dose adjustments. 19 Furthermore, few treatment options are available for patients with end-stage renal disease. Indeed, current recommendations only allow the use of grazoprevir (NS3/NS4A inhibitor) and elbasvir (NS5A inhibitor) in combination in these patients.25,26 Despite overall promising results, the optimal DAA combination, the treatment duration and treatment dosages in these special populations require further investigations.
Limitations in the application of DAA regimens also include certain viral genotypes. HCV genotype 3 is considered to be the most difficult-to-cure genotype with DAA-based therapy.27,28 Currently available DAAs are less efficient against this genotype. Two regimens are recommended for the management of these patients: the combinations of daclatasvir and sofosbuvir or sofosbuvir and velpatasvir. The SVR rates are lower compared with other genotypes, ranging from 50% to 90% depending on the treatment history and the disease stage.27–31 Interestingly, HCV genotype 3 is the second most prevalent genotype worldwide with high morbidity and mortality rates compared with other genotypes. 32 Therefore, additional studies are needed to improve new therapeutic options for patients with HCV genotype 3 infection.
Another aspect that requires further studies is the persistent risk of HCC post cure in patients with advanced fibrosis or comorbidities. Although the risk of de novo HCC development is reduced after SVR, HCC can occur even more than 10 years following successful HCV clearance.4,33,34 Evidence from several cohorts appears to suggest that post-SVR HCC development and recurrence may be more frequent after DAA treatment compared with IFN-based therapy, potentially due to a difference in host immune modulation between IFN- and DAA-treated patients.4,35 Given the increasing incidence of HCC and the urgent unmet needs for prevention and detection of HCC, alternative strategies for these populations should be explored.7,34
Although not an issue in the majority of treated patients, HCV resistance to DAAs can occur in a small minority of patients. HCV has a quasispecies distribution. The high replication rate of HCV (1010−1012 virions per day in patients with chronic infection) and the low fidelity of its RNA polymerase (1.5–2.0 × 10−3 base substitutions per genome site per year) result in a high degree of genetic variants. Therefore, patients are infected by a mix of distinct but closely related viral populations. 36 Viral polymorphisms can naturally appear in regions targeted by DAA and may confer DAA resistance. When a DAA treatment is administered, sensitive wild-type populations are completely inhibited while drug-resistant variants are rapidly selected, leading to treatment failure (breakthrough) or relapse after therapy.17,37 Moreover, DAAs from the same class share cross resistance, meaning that specific mutations can confer reduced susceptibility to all molecules from the same class, thereby limiting retreatment options.17,38 Consequently, more studies and clinical trials are ongoing to define the best retreatment strategies for patients with viral resistance or treatment failure.
Targeting of viral host-dependency factors to prevent and cure HCV infection
The HCV life cycle can be divided into three major steps: viral attachment and cell entry; viral translation and replication; and assembly and release of the new infectious virions (Figure 1). 1 Numerous host cell factors are involved in each step of the HCV life cycle. A hallmark of HCV is the association of circulating viruses with very-low-density and low-density lipoproteins (VLDL and LDL) forming infectious lipo-viro particles (LVPs). 39 HCV infection of hepatocytes begins with a complex interaction between these LVPs and several cellular attachment or entry factors. After attachment, viral particles are then internalized through clathrin-mediated endocytosis. 40 The viral RNA is released into the cytosol, and translated into a polyprotein that is targeted to the endoplasmic reticulum (ER) where it is subsequently processed by host and viral proteases to generate three structural proteins (Core, E1 and E2) and seven NS proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B). 41 The NS proteins form the replication complex at the surface of ER-derived membranes, the so-called membranous web. Viral replication is catalyzed by the viral polymerase NS5B. 42 Viral RNA and proteins accumulate and new virions are assembled in an ER-related compartment in close vicinity to hepatic lipid droplets (LDs). Finally, HCV uses the VLDL production and secretory pathways to generate and export infectious LVPs. 43 As reviewed below, many of these host-dependency factors are targets for antiviral drugs. This review will focus on host factors that have been explored as targets of HTAs in antiviral therapies.
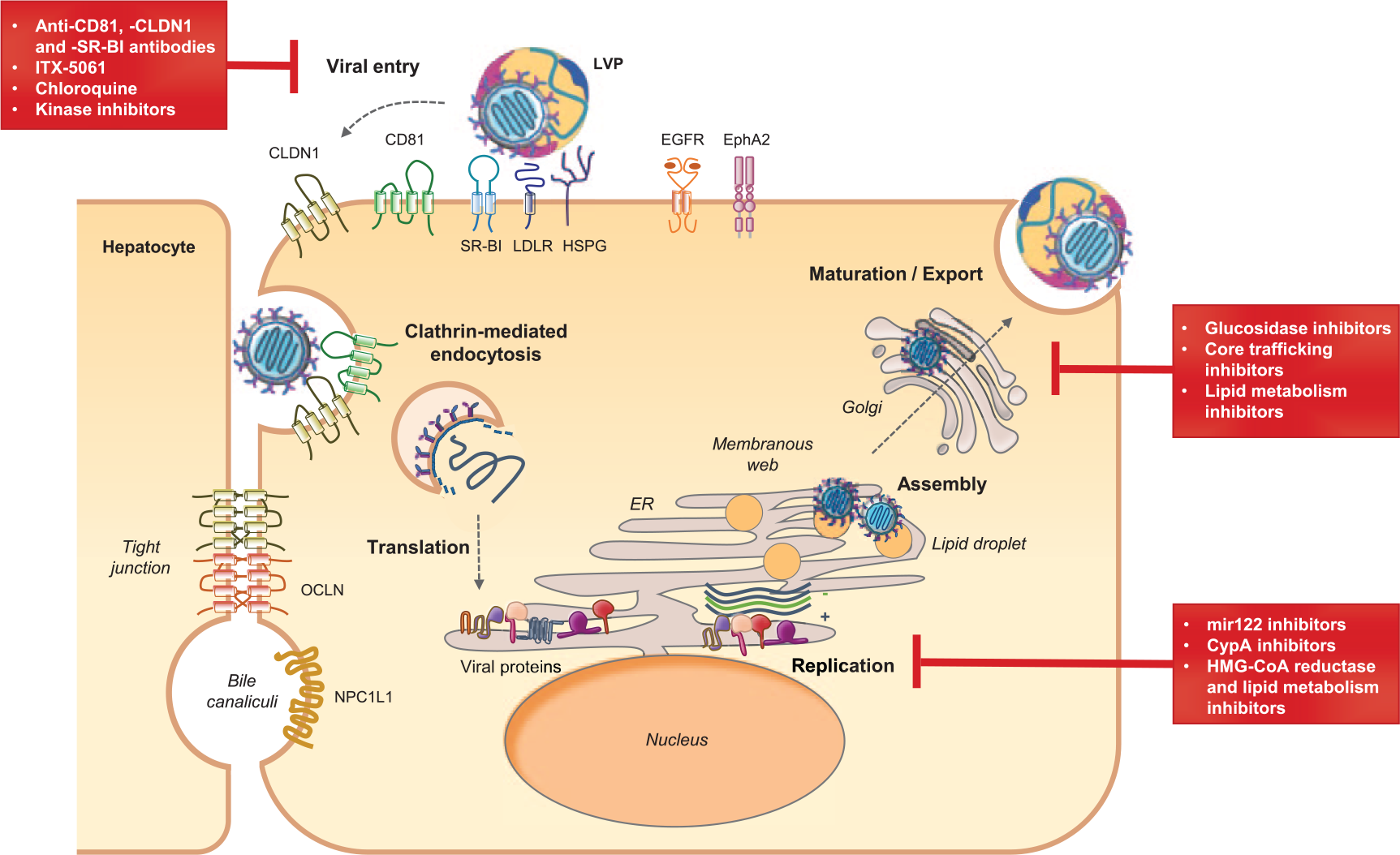
The HCV life cycle and host targets for antiviral therapy.
Inhibitors of HCV entry: prevention of HCV infection during transplantation
The HCV entry process has been particularly well characterized in the past few years. First, heparan sulfate proteoglycans (HSPGs) are involved in the attachment of viral particles to hepatocytes. Viral entry is then mediated by several entry factors. The main entry factors are the scavenger receptor BI (SR-BI), the tetraspanin CD81 and the tight-junction proteins Claudin 1 (CLDN1) and Occludin (OCLN). 44 This process also involves numerous cofactors, notably the two receptor tyrosine kinases epidermal growth factor receptor (EGFR) and ephrin receptor A2 (EphA2) that promote CD81–CLDN1 interaction, as well as other tetraspanin-associated proteins.45,46 Finally, other regulators of the host lipid metabolism and innate immune responses such as the Niemann-Pick C1 Like 1 protein (NPC1L1) and the sodium taurocholate cotransporting polypeptide (NTCP) are required for HCV entry.44–48 Furthermore, transferrin receptor 1 (TfR1) has recently been proposed as an HCV entry factor. 44
Targeting host proteins involved in virus entry is a particularly attractive strategy for the prevention of organ infection in LT or during transplantation of HCV-positive organs, as reviewed recently.49,50 Chronic HCV infection is a leading cause of LT. Infection of the engrafted organ is universal and patients exhibit accelerated progression to advanced liver disease following LT.49,50 A very appealing option to prevent HCV infection and viral-induced disease in the graft is blocking viral cell entry using entry inhibitors alone or in combination with other antiviral treatments.
The first HTA having entered clinical development is the small molecule ITX-5061 that blocks the interaction of HCV with SR-BI. In vitro studies have shown efficient reduction of virus entry by ITX-5061 for HCV genotypes 1–6. 51 Although SR-BI is a key component of the host lipid metabolism, ITX-5061 was well tolerated in clinical phase I studies with the only major adverse effect being elevated serum levels of high-density lipoproteins (HDLs). 52 While ITX-5061 showed only limited efficacy in clinical phase I studies enrolling patients with chronic HCV infection, it significantly limited viral evolution in patients undergoing LT.52,53 Of note, long-term treatment with ITX-5061 in cell culture resulted in escape mutations in HCV envelope glycoprotein E2, but these findings might be limited to in vitro settings, since the mutants also showed increased sensitivity to antibody neutralization and thus might not arise in vivo. 54 Limited potency of the compound combined with viral escape may have been the reasons for incomplete protection.
Another attractive strategy to inhibit virus entry is to target host factors using specific monoclonal antibodies. These antibodies bind to host proteins and block their engagement by HCV, thereby blocking virus entry into hepatocytes. Antibodies targeting CD81, CLDN1 and SR-BI have been shown to elicit strong antiviral effects in preclinical mouse studies.55–60 Of note, anti-CLDN1 antibodies are also able to cure chronic HCV infection in the albumin enhancer-promoter-driven urokinase-type plasminogen activator/severe combined immunodeficiency (uPA/SCID) liver chimeric mouse model, suggesting that entry inhibitors also hold potential to treat chronic hepatitis C.59,60 Completion of preclinical development is the next step for clinical translation of these compounds. Further host factors involved in HCV entry that are subject to (pre-)clinical drug development include NPC1L1 and EGFR. Ezetimibe, a molecule targeting NPC1L1, elicited only minor effects on HCV viral loads in a phase I clinical trial that enrolled two patients who underwent organ transplantation. 61 Additionally, a phase I/II clinical trial has been initiated to evaluate the potential of the clinically approved EGFR inhibitor erlotinib to treat chronic HCV infections [ClinicalTrials.gov identifier: NCT01835938].
Silymarin/silibinin is a clinically approved natural product isolated from milk thistle that is regularly used to treat liver damage after intoxications and chronic liver disease. In vitro studies have shown an inhibitory effect of this compound on clathrin-dependent viral trafficking. 62 Clinical phase I and phase II studies of patients with chronic HCV, including those who have had LT, revealed conflicting results. While some studies reported a significant reduction in viral loads and alanine transaminase (ALT) levels, other studies reported viral rebound and treatment failure.63–66 Further studies are thus needed to clarify the potential of silymarin/silibinin for the management of chronic hepatitis C.
Another drug targeting virus entry that has been evaluated in a phase IV clinical study is the malaria drug chloroquine that affects clathrin-mediated endocytosis and virus-mediated autophagy.67,68 Monotherapy with chloroquine of patients with chronic HCV infection not responding to IFN-based regimens with chloroquine resulted in a significant reduction of viral RNA and liver ALT levels. However, viral titers quickly relapsed after cessation of treatment, indicating that monotherapy with chloroquine might not be sufficient to cure HCV infection. 69
Interestingly, combinations of host-targeting HCV entry inhibitors and DAAs are characterized by synergistic antiviral effects.58,70 The prophylactic properties of HTAs targeting virus entry could make them a valuable asset in the prevention of graft infection during LT. Additional clinical trials are required to establish the place of entry inhibitors in the management of HCV graft infection.
Inhibitors of HCV RNA replication
Cyclophilin A inhibitors
To facilitate its replication, HCV uses a plethora of host factors, including cyclophilins, that interact with the viral protein NS5A.42,71–73 It was demonstrated that cyclosporine A (CsA), a common immunosuppressive drug targeting cytochrome P450 A (CypA), efficiently suppresses viral replication in vitro and in LT recipients.74,75 Based on these observations, CsA derivatives lacking immunosuppressive activity but retaining antiviral activity have been developed.76–78 Three molecules have so far exhibited clinical efficacy in patients with HCV infection treated with both IFN-based and IFN-free regimens: alisporivir/Debio 025, NIM811 and SCY-635.79,80 Safety limitations have delayed the clinical development and further studies are needed to investigate the role of these compounds in the management of patients with HCV infection. These compounds act through two distinct mechanisms. On one hand, they inhibit HCV replication by disrupting and preventing Cyp–NS5A interaction. On the other hand, CypA inhibitors can restore the host innate immune responses against HCV.79,81,82 Interestingly, it was demonstrated that alisporivir also restricts human immunodeficiency virus (HIV) replication in vitro and in vivo by inhibiting CypA–HIV capsid protein interaction.83–85 Based on this knowledge, Gallay and collaborators described a novel CypA inhibitor, named CPI-431-32, that simultaneously blocks HCV and HIV replication with a higher efficiency than alisporivir. 86 CypA inhibitors may thus be of interest to treat patients with HIV/HCV coinfection.
MicroRNA-122 inhibitors
After viral entry, the stability of HCV RNA and its propagation in hepatocytes depend on the interactions between the HCV genome and microRNA-122 (miR-122), a miRNA highly expressed in the liver.87–89 It was demonstrated that sequestering miR-122 using miravirsen/SPC3649, a miR-122 antisense locked nucleic acid, strongly reduces HCV replication in vitro.87,90 Moreover, as the miR-122 binding sites are highly conserved across HCV genotypes, miravirsen exhibits a pan-genotypic antiviral effect. 91 It also appears safe and well tolerated and elicits a prolonged dose-dependent reduction in HCV RNA levels in patients without serious adverse effects.92,93 Although both in vitro and clinical data indicated that miravirsen provides a high barrier to viral resistance, some patients experienced viral rebound within 14 weeks after treatment cessation, suggesting that miravirsen may not be used as a monotherapy but rather in combination with other HTAs or DAAs.91,92 In this respect, promising results have already shown that miravirsen and DAAs exert an additive effect in vitro and that miravirsen is fully active against DAA-resistant HCV variants.74,94 Finally, a recent phase I clinical study assessed the safety and the antiviral effect of RG-101, a hepatocyte-targeted N-acetylgalactosamine-conjugated oligonucleotide that antagonizes miR-122. A single dose of this compound resulted in a significant decrease in HCV load and was well tolerated in patients with chronic HCV infection with various genotypes. However, viral rebound was observed in two patients, likely due to substitutions in miR-122 binding sites in the HCV genome, which led to viral resistance. 95 More studies are therefore needed to evaluate the potential of these antiviral strategies. A phase II study is now underway (EudraCT 2015-001535-21) to establish the efficacy of RG-101 in combination with DAAs. Since HTAs target host factor functions and not the virus itself, these compounds may have adverse effects linked to their mode of action.3,83,96 Thus, although no side effects have been observed so far after administration of miR-122 inhibitors, the effect of a long-term use of such compounds must definitely be investigated because low hepatic miR-122 levels have been associated with liver disease progression and HCC development.97–99
Inhibitors of lipid biosynthesis pathways
Statins are drugs widely used for the treatment of hypercholesterolemia. They act through the inhibition of the 3-hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol biosynthesis in the liver. HCV replication can be disrupted in vitro by several statins such as lovastatin, atorvastatin, fluvastatin and simvastatin.100–102 Thus, statins have been proposed as candidates for the treatment of HCV infection. Of note, all HMG-CoA reductase inhibitors do not affect HCV replication, such as apravastatin, which does not exhibit an anti-HCV activity. 103 The precise mechanism of action is not yet fully understood. Some studies suggested that the anti-HCV activity of statins could be due to the inhibition of geranylgeranylation of cellular proteins rather than to the inhibition of cholesterol biosynthesis.100,104 Geranylgeranylation is a post-transcriptional modification that attaches geranylgeranyl groups produced through the cholesterol biosynthesis pathway to host proteins to facilitate their association with the host-cell membrane, which is essential for viral replication. 105 Initial clinical studies indicated that statin monotherapies do not significantly modulate viremia in patients with chronic infection.106–108 Broad retrospective studies subsequently demonstrated that statins constitute interesting adjuvants for anti-HCV therapies. Fluvastatin and pitavastatin have been reported to increase SVR in patients with HCV infection treated with pegylated IFNα and ribavirin.109–113 Moreover, statins have been suggested to offer chemoprevention against HCV-induced HCC by inhibiting cell growth and tumor spread, and by exerting immunomodulatory effects.114–116 The use of statins as adjunctive therapy in HCV treatment has been limited to IFN-based therapies. Although statins in combination with DAAs have been shown to increase antiviral efficacy against HCV infection in vitro, the clinical benefit of statins in the era of DAA is still uncertain. Moreover, concerns have been raised about possible statin–DAA interactions.102,117–119 Therefore, future studies are needed to better characterize the benefits of statins in anti-HCV therapies and for the prevention of HCC development.
Another example of a lipid biosynthesis pathway inhibitor is PF-429242, which inhibits the human subtilase SKI-1/S1P. SKI-1/S1P regulates a master lipogenic pathway upstream of HMG-CoA reductase through the activation of the sterol-regulating element binding protein. It was shown that the antiviral activity of PF-429242 was higher than that of statins in cell culture and that this compound decreased virion production in vitro.120,121 However, their suitability for antiviral treatment still needs to be investigated.
Inhibitors of HCV assembly and release
α-Glucosidase inhibitors
The HCV particle consists of a nucleocapsid containing the viral RNA, surrounded by an ER-derived membrane where E1 and E2 glycoproteins are anchored as heterodimers. 43 In the ER, E1 and E2 proteins are highly glycosylated by α-glucosidases I and II, which ensure proper folding of these proteins. Celgosivir, a glucosidase I inhibitor, was shown to reduce virion production and viral infectivity in vitro.122,123 It also exhibited antiviral activity in preclinical trials and was investigated in a phase II clinical trial. Although this compound is not efficient as a monotherapy, it demonstrated a synergistic effect in combination with IFN-based therapies [ClinicalTrials.gov identifier: NCT00332176]. 122 The clinical trials for celgosivir were stopped as stated in the Migenix financial report for 2010.
Inhibitors of HCV core protein trafficking
Since HCV circulates as a LVP in the blood of patients with infection, viral assembly and egress of infectious LVP rely on host factors, which are required for lipid metabolism and VLDL production. Therefore, they represent potential targets for anti-HCV therapies.39,124,125 Virus assembly is triggered by core protein recruitment on LD by the diglyceride acyltransferase I (DGAT-I).126,127 It was demonstrated that DGAT-I inhibitors efficiently suppress production of infectious viral particles in vitro while LD formation is not affected. Interestingly, quercetin, a natural flavonoid that inhibits DGAT-I, was reported to have anti-HCV properties. 128 In a phase I dose-escalation study, quercetin exhibited both high safety (up to 5 g/day) and high antiviral efficacy in patients with chronic infection. 129 Quercetin is widely available and cheap, and could thus be developed as an inexpensive adjuvant for HCV treatment. Recently, the antiviral efficacy of the DGAT-I inhibitor LCQ908/pradigastat was assessed in phase II clinical trials in patients with HCV infection. Pradigastat was also safe and well tolerated. However, the clinical study was prematurely interrupted for lack of efficacy: no significant change in serum viral RNA levels was observed in patients after pradigastat treatment compared with the placebo group. 130 More studies are now needed to determine whether the DGAT-I inhibitor could be beneficial for patients in combination with DAAs.
During HCV assembly, HCV core protein is recruited from LD to the viral assembly site. This process involves several host factors, including clathrin assembly protein complex 2 medium chain μ1 (AP2M1), which directly interacts with core protein. Furthermore, the adaptor-associated kinase 1 (AAK1) and the cyclin-associated kinase (GAK) are known to regulate core-AP2M1 interaction and are essential for HCV assembly.131–133 Accordingly, Neveu and colleagues discovered that AAK1 and GAK inhibitors, including the approved anticancer drugs sunitinib and erlotinib, can block HCV assembly.131,132 However, these compounds were initially developed to target other kinases and could have adverse effects due to their lack of specificity. To overcome this problem, a more specific GAK inhibitor, isothiazolo[5,4-b]pyridine, was recently developed. This new drug efficiently inhibits HCV entry and assembly in vitro with limited off-target effects and has been proposed as an antiviral strategy. 133
Inhibitors of host lipid metabolism
During the later stages of assembly, HCV coopts the VLDL pathway.124,134 Formation of VLDL as well as LVP requires the microsomal triglyceride transfer protein (MTP). This host enzyme mediates triglyceride incorporation into nascent LD and allows lipid loading of ApoB in the ER. Compounds that inhibit the VLDL assembly pathway, such as MTP inhibitors or ApoB inhibitors, are therefore interesting candidates to block HCV assembly and release.124,134 In line with these observations, the MTP inhibitor naringenin, a grapefruit flavonoid, has been shown to inhibit VLDL secretion in vitro and in vivo as well as HCV secretion in cell culture. 135 More recently, a study demonstrated that the MTP inhibitor amiodarone also downregulates HCV assembly and release. 136 However, a recent report indicated that amiodarone could induce bradycardia in patients when administered in combination with DAAs. 96 Several other MTP inhibitors are currently being evaluated in clinical trials for the treatment of dyslipidemia but their in vivo efficacy against HCV remains to be demonstrated.137–139 Interestingly, mipomersen, an antisense inhibitor of ApoB synthesis used for the treatment of hypercholesterolaemia was also shown to efficiently block HCV morphogenesis in vitro. 140 Mipomersen now needs to be tested in vivo.
In 2011, Goldwasser and colleagues showed that naringenin blocks HCV assembly not only by inhibiting MTP but also by activating the peroxisome proliferator-activated receptor α (PPARα) in HCV-infected cells.141,142 PPARα is a nuclear transcription factor regulating several aspects of the lipid metabolism in the liver. Notably, its activation leads to increase of fatty acid oxidation and impairment of HCV assembly and release.143,144 During chronic HCV infection, the viral core protein inhibits PPARα activity to promote HCV replication. Thus, restoring PPARα activity constitutes an interesting strategy to inhibit HCV infection.145,146 Accordingly, several approved drugs targeting PPARα were shown to display antiviral activity against HCV. This holds true for resveratrol and its methylated form pterostilbene (two natural compounds extracted from grapes and blueberries) as well as for torimefene (a tamoxifene derivative).146,147 In addition, Fujita and collaborators demonstrated the efficacy of bezafibrate, another PPARα agonist commonly used against hyperlipidemia in patients with chronic HCV infection. 148 However, in 2013, Knop and colleagues observed that the beneficial effect of bezafibrate in patients is not due to a decrease in HCV RNA levels but rather to a significant reduction of liver enzymes and improved liver function in patients. 149 Finally, more recent studies corroborated the efficacy of bezafibrate in the regulation of lipid metabolism and in the reduction of viral loads in cell culture, indicating that further studies are needed to ascertain its potential for anti-HCV therapies.150,151 Clinical trials are needed to investigate the clinical antiviral efficacy of assembly inhibitors.
Future role of HTAs in HCV therapy
DAA-based treatment is standard of care for the management of patients with chronic HCV infection. Next-generation DAAs with an even higher barrier to resistance and pan-genotypic activity are currently under clinical development. 3 In this context, one can wonder about the positioning of HTAs in anti-HCV therapy. A key advantage of HTAs could be linked to the question of whether next-generation DAAs can address the limitations of currently licensed combination therapies (patients who are difficult to treat, resistance, access and HCC risk). While it is expected that next-generation DAAs will indeed address several of these issues, it is likely that DAAs will not be able to address all of these remaining challenges.
Multiresistance, especially in patients with reinfection such as those who are drug abusers with limited compliance and have had multiple treatment courses, may require complementary compounds. This is already blatant for nosocomial bacterial infections in patients who are critically ill, in whom current antibiotics are no longer effective. HTAs may also prove useful to ameliorate current treatment approaches when combined with DAAs. Combinations of HTAs and DAAs may even further reduce treatment duration, increase efficacy and thus improve adherence and access to therapy. HTAs may also be used for the treatment of patients with advanced disease, to lower HCC risk since this is a limitation of current DAA regimens, which have been proven unable to prevent HCC, especially in patients with advanced fibrosis or comorbidity. Finally, host-targeting entry inhibitors are good candidates to prevent HCV infection during LT or transplantation of HCV-positive organs such as kidneys. It is clear that infection prevention is by far conceptually a better option than DAA treatment post transplantation, since if the infection develops, it entails the risk of acute or chronic HCV-induced liver disease (with fibrosing cholestatic hepatitis or HCC as the most severe forms). If a short-term preventive approach can effectively prevent HCV infection during transplantation, this concept may also improve patient care as it will be administered perioperatively during hospitalization.
Taken together, further clinical trials are needed to define the place of HTAs in the management of patients with HCV infection and to determine their role in comparison to or in combination with DAAs.
Footnotes
Acknowledgements
The authors acknowledge Dr Marie Meister for critical reading of the manuscript.
Funding
The authors acknowledge funding by ARC, Paris and IHU Strasbourg (TheraHCC IHUARC IHU201301187), the European Union (ERC-AdG-2014-671231-HEPCIR, EU H2020-667273-HEPCAR, FP7 HEPAMAB GAN 305600), ANR (LABEX ANR-10-LABX-0028_HEPSYS), the National Institutes of Health (1U19AI123862-01) and the DoD (CA150281)
Conflict of interest statement
C.S., M.B.Z. and T.F.B are inventors of patents or patent applications on anti-receptor antibodies for prevention and treatment of HCV infection.